Overcoming Bottlenecks in AAV Manufacturing for Gene Therapy
Cell Gene Therapy Insights 2018; 4(8), 815-827.
10.18609/cgti.2018.083
Recombinant adeno-associated viral vectors (rAAV) have emerged as one of the most powerful tools for gene delivery to treat human disease. Significant advancements in vector development and manufacturing methods, along with promising results in the clinic, has initiated an extraordinary interest in drug development for gene therapies and commercialization. It is an exciting time for AAV-mediated gene therapy; in 2012, the field witnessed the ap-proved use of the gene therapy product, Glybera, for the treatment of lipoprotein lipase deficiency by the European Medicines Agency, and in 2017, the approval of Luxturna, for the treatment of Leber’s Congenital Amaurosis by the US FDA, thus arriving at the threshold of gene therapies reaching patients. Still, with an increasing number of gene therapy programs and a growing de-mand for both pre-clinical and clinical grade vector, the industry must be prepared to implement practical solutions to address the manufacturing bottlenecks and rigorous product characterization needed to meet higher regulatory requirements earlier in clinical development in order to efficiently advance and market these products. Below is an overview of the current status of rAAV vec-tor production from academic to contract manufacturing organization (CMO).
Submitted for Peer Review: 23 Aug 2018 Published: 6 Nov 2018
Introduction
AAV Virology
AAV belongs to the parvovirus family, specifically the dependoparvovirus genus. Parvoviruses are among the smallest of the DNA viruses at approximately 25 nm in diameter. Members of the dependoparvovirus genus require co-infection with a helper virus, such as adenovirus (Ad), herpes simplex virus or vaccinia virus to allow productive infection and replication. The wild-type AAV genome is a linear, single-stranded DNA molecule consisting of three open reading frames encoding four replication proteins (Rep 78, 68, 52, 40), three capsid proteins (Cap) VP1, VP2, VP3, and the assembly activating-protein (AAP), all of which are flanked by two 145bp inverted terminal repeats (ITR) [1,2]. Samulski et al. described the first cloning and rescue of AAV from plasmid DNA in human cells [3], and the first use of a rAAV vector to express foreign genes in mammalian cells was described by Hermonat and Muzyczka [4]. Early production of rAAV vectors required the use of auxiliary helper viruses, such as Ad, along with the transient co-transfection of two plasmids: one carrying the vector plasmid encoding the transgene of interest (GOI) flanked by the ITRs, and the second plasmid containing the AAV Rep, Cap and AAP functions.
To date, there are four scalable production platforms that have been established to generate rAAV vectors:
- Transient transfection of plasmid DNA in mammalian cells [5,6];
- Adenovirus (Ad) infection of stable mammalian cell lines containing both AAV rep/cap and therapeutic rAAV genome [7,8] as well as co-infection of mammalian stable cell lines containing AAV rep/cap with Ad and rAd-AAV hybrid virus [9];
- Recombinant baculovirus infection of insect cells [10–16]; and
- Infection of mammalian cells with two recombinant herpes simplex virus (rHSVs); one containing the therapeutic rAAV genome (rHSV-AAV) and the second containing AAV rep/cap (rHSVrepcap) [17,18]
All of the above have been recently reviewed by Penaud-Budloo et al. [19]. Here we will provide a brief overview of the two most common methods used to produce clinical grade rAAV as well as discuss several of the challenges faced in vector manufacturing and scale-up.
The viral vector manufacturing process is complex. Significant investments in time, expertise and money are required to meet the growing demand and quality. Some of the significant challenges facing manufacturers include but are not limited to: (1) developing scalable technologies to produce vectors at acceptable costs while transitioning from pre-clinical to clinical development, and ultimately, commercial stage manufacture; (2) understanding and maintaining product quality throughout the manufacturing process; (3) meeting new regulations as the industry matures; and lastly, (4) balancing high product quality and safety with accelerated regulatory pathways.
Transient transfection
Transient transfection of plasmid DNA into cell lines was the first, and remains to be, the most common method for pre-clinical and clinical grade manufacturing of rAAV. A major improvement to the transient production system was the identification of the critical adenoviral genes required for rAAV replication (E1A/B, E2A, E4, and VA RNAs), and their cloning into an Ad helper plasmid [5,20]. The most established protocol is the co-transfection of the vector plasmid, containing the therapeutic GOI, along with equimolar amounts of one or two helper plasmids into human embryonic kidney (HEK) 293 cells through condensation with polyethyleneimine (PEI) or calcium phosphate. The helper plasmid(s) provide the AAV Rep, Cap and AAP functions, as well as the necessary adenoviral genes required for rAAV replication and encapsidation. The fourth adenoviral function required for AAV replication, E1A/E1B, are constitutively expressed in HEK293 cells [21]. Alternatively, early genes from the herpes virus can be used instead of the Ad helper [22]. The first large scale production of rAAV using HSV helper was clinically tested by Solid Biosciences. Although clinically managed, unexpected laboratory findings were reported in the first patient dosed. The event was unexpected and therefore, classified as a suspected unexpected serious adverse reaction (SUSAR), resulting in FDA clinical hold that remains to be determined. Due to the timing of the event being “several days after administration,” it is believed to be production related [23]. The most widely used protocol, the triple plasmid transfection, consists of two helper plasmids separating the Rep/Cap functions from those of the Ad, offers flexibility to rapidly switch from one serotype to another by simply changing the Rep/Cap plasmid. Plasmids are typically manufactured using standard techniques in E. coli including bacterial origin and antibiotic- resistance genes, by minicircle (MC) technology [24,25] or by Doggybone technology [26] wherein the latter two substrates offer certain regulatory advantages due to the removal of bacterial plasmid backbone sequences. This concern has become more important since the Sarepta AAV gene therapy program to treat Duchenne muscular dystrophy (DMD) was also placed on clinical hold for a production related issue described later in the text [32]. Although transient transfection in adherent HEK293 cells has been used for clinical manufacturing of rAAV vectors, there is a fundamental lack of scalability due to the large number of flasks, roller bottles, cell stacks, or otherwise, fixed-bed culture vessels, required for a clinical production run. Fixed-bed bioreactors, such as the iCellis™ (Pall) bioreactor, allow increased culture surface areas within a single vessel while offering for some degree of in-process control by maintaining sufficient gas exchange, nutrients, as well as reducing the accumulation of deleterious by-products of cell culture such as ammonia and lactic acid. Nonetheless, these systems are limited in surface area, the cell densities that can be achieved, and ultimately, require additional scale-out for increased product [28]. These campaigns are often lengthy, utilize animal derived components, cost-ineffective and require multiple production batches to meet the needs of the clinical trial. Suspension-adapted HEK293 cells, on the other hand prove to be scalable from bench to commercial scale stirred-tank bioreactors, and conditions to be economically viable in the long term [6].
Grieger et al. at the University of North Carolina Gene Therapy Center developed a scalable, transfection-based manufacturing process utilizing suspension HEK293 cells to address the above stated challenge. Adherent HEK293 cells from a qualified clinical master cell bank were adapted to grow in suspension in an animal-derived component free and antibiotic-free media, in shake-flasks, WAVE bag bioreactors and stirred-tank bioreactors. The use of animal-derived component free media is a significant regulatory advantage as it reduces the risk of introducing adventitious agents, simplifies purification and ultimately increases product safety. The yields generated utilizing the triple plasmid transfection are typically greater than 105 vector-genome containing particles per cell (vg/cell) in crude lysates or greater than 1 × 1014 vg/L of cell culture (1 × 106 cells/mL from 0.1 – 20L suspension) after 48 hours incubation [6]. Parameters including selection of a compatible serum-free, chemically defined suspension media optimal for both cell growth and transfection, selection of a transfection reagent, transfection conditions and cell density, were optimized. Linear increases in rAAV yields with respect to cell density are observed, achieving pre-purification yields of ~1 × 1015 vg/L of high-density cell culture in both shake-flasks and stirred-tank bioreactors up to 250L scale [Smith JM, Unpublished Data]. Increased yield recovery using improved purification methods will achieve >4 × 1014 purified AAV vector per L of transfected higher cell density culture. This platform has all the pre-requisites to enable rapid and scalable rAAV production for larger-scale manufacturing to support Bio/Tox, GMP Phase 1/2 and commercial campaigns. Baxter/Shire, Pfizer and Viralgen have invested in the triple transfection process of suspension HEK293 cells (Asklepios Biopharmaceutical, Inc Pro10™ cells) for both clinical and commercialization of rAAV vectors [29–31].
Transient viral vector production through the use of plasmid DNA is a complex materials supply chain, with the plasmid DNA constructs used as a starting material at the front of the line. For the generation of GMP source plasmid starting material, it takes approximately 6 weeks to produce a GMP-Source bacterial master cell bank (MCB) followed by approximately 6–8 weeks for GMP-Source plasmid manufacturing and QC release testing (Figure 1B). Of course, the latter varies slightly depending on the amount of plasmid needed to sufficiently generate enough rAAV vector to cover the patient dose requirements for the clinical trials. It’s no surprise that as the manufacturing platforms scale up, the amount of plasmid required may increase proportionally. The use of non-GMP grade plasmids for use as starting materials for manufacturing rAAV for early-stage clinical trials has currently been allowed. Due to trace amounts of non-product specific DNA found in the plasmid DNA starting material that lead to Sarepta’s clinical hold, it is plausible that the associated quality requirements for plasmid DNA manufacturing could be increased, requiring the use of GLP or GMP grade plasmid DNA for rAAV manufacturing [32].
Although the steady supply of plasmid DNA is both a costly and timely investment, gene therapy manufacturers can overcome the bottleneck of long lead times by aligning with suppliers capable of large-scale GMP or GMP-Source plasmid manufacture. Early yield assessment and optimization of AAV helper and transgenic vector plasmids should be performed alongside the generation of GMP bacterial master cell banks. In doing so, manufacturers can secure a timely and steady supply of plasmid to support early clinical through commercial production. Alternatively, novel technologies such as Doggybone DNA (dbDNA), produced using in vitro, bacterial-free, enzymatic methodologies, may address many of the challenges of plasmid DNA production including scalability, high costs and long lead times.
Infection with recombinant baculovirus
While vector suspension cell-based production methods utilizing the triple plasmid transfection are one strategy, the baculovirus-Sf9 platform has been notably established as a cGMP-compatible and scalable system (>200 L) capable of producing as many vector genomes per cell in the crude harvest as the mammalian cell-based transfection methods (i.e., ~1-2 x 105 vg/cell) [10].
Originally developed by Urabe et al., suspension-adapted insect cells from Spodoptera frugiperda are cultured using serum-free medium and can be infected by one, two, or three baculovirus expression vectors (BEV) that provide the necessary helper functions to promote rAAV production [11]. Optimizations from Smith et al. led to the development of a process reducing the number of recombinant baculoviruses from three to two: a recombinant BEV providing Rep78/52 and Cap followed by a second recombinant BEV carrying the transgenic vector flanked by the AAV ITRs [12]. To date, the baculovirus-Sf9 platform has been modified to require the infection of only a single recombinant BEV through the use of stably transformed Sf9 cell lines expressing the Rep and Cap proteins [13,14]. However, the dual BEV system is the version of this platform that provides the most flexibility and is widely used to generate rAAV vectors. In general, the baculovirus-Sf9 system has demonstrated production capabilities up to >1016 vg from 200 L stirred-tank bioreactors [15].
From a regulatory perspective, the baculovirus-Sf9 production system has some advantages over other production platforms regarding safety. Some of which include, the use of serum-free media [16], besides baculovirus, no additional helper virus is required for rAAV production, and most insect viruses do not actively replicate in mammalian cells; although, there have been adventitious virus transcripts observed in Spodoptera frugiperda cell lines [33,34]. Many of the process-related impurities require more extensive characterization of baculovirus and Sf-derived contaminants (i.e., residual DNA, protein, etc.) as seen in the EMA assessment report of the aforementioned product, Glybera [27].
Like the transient transfection system, the baculovirus-Sf9 platform has a complex materials supply chain. Even with the single BEV production system, multiple cloning steps, followed by transfection, BEV(s) high producing clone selection and amplification are required to generate the BEV master and working viral banks to be used as starting material (Figure 1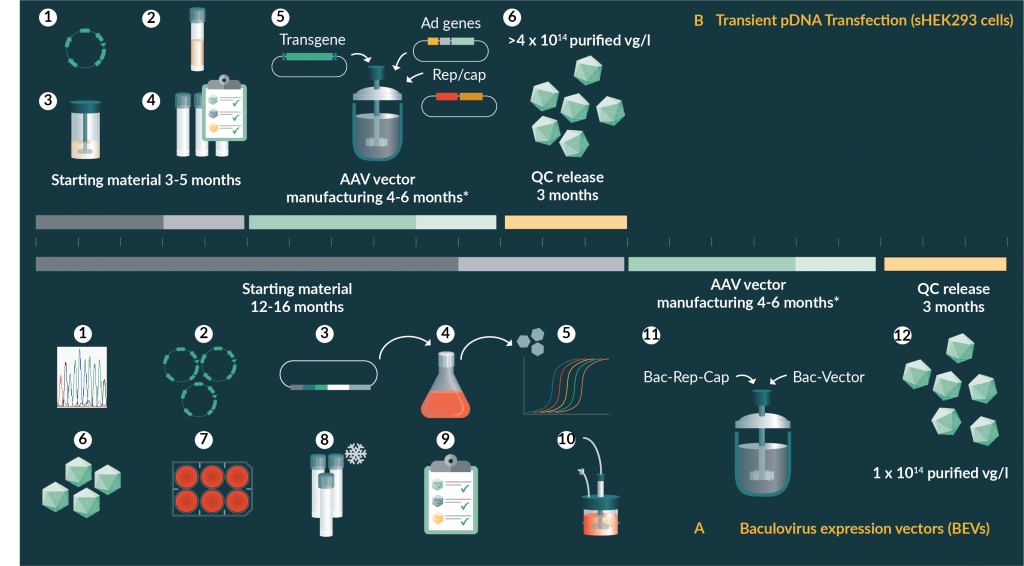
Regardless of the final rAAV production platform used, the triple plasmid transfection system is used by all groups for screening, optimization, and identification of early stage clinical rAAV candidates due to the speed, and overall, lower initial investment. Manufacturers utilizing the baculovirus-Sf9 platform should have a greater understanding of product attributes to strengthen comparability when transitioning between manufacturing platform technologies from early to late stage.
Manufacturing scale-up
Manufacturing scale-up is a critical step in the advancement of therapeutic drug products. Because gene therapies have the potential to deliver life-saving therapies to patients, clinical program development timelines may be accelerated by regulatory agencies. As a result of this, as well as external competitive programs, manufacturing timelines could become compressed forcing development teams to lock-in process steps during early stages of the program making it more difficult to implement process improvements as well as launch to commercial scales capable of producing vectors at the right quantity, quality and cost. In general, manufacturers are faced with the option to either scale up, in a vertical fashion, by increasing the size of one or more unit operations, for example, increasing the size of the bioreactor, or scale out, in a horizontal fashion. The former is typically the first approach as it calls for a similar workforce with relatively little impact to the overall manufacturing run rate. For many gene therapies, however, the accelerated timelines, limited process characterization and overall lack of scalability, drive manufacturers to scale out. Transient transfection processes utilizing adherent producer cell lines often succumb to this reality.
Often manufacturers must provide a steady supply of material for ongoing clinical trials, while concurrently working to develop and improve manufacturing processes. For this reason, it’s critical to design meaningful experiments evaluating fundamental principles of scale-up. In the case of upstream manufacture, assessing scale-dependent parameters related to bioreactor mixing characterization: power input per unit volume, bulk mixing time, volumetric gas (O2) transfer (kLa), and tip speed (shear), as well as those factors directly related to cell culture (pH, temperature, dissolved oxygen [DO]) and vector production technologies. It is also necessary to appropriately stage experiments based on available screening methodologies and the reliability of the test model. For example, small-scale shake-flask studies can be conducted to evaluate critical factors in the transient transfection of mammalian cells to produce rAAV such as transfection reagent, plasmid DNA concentration, ratio of DNA: transfection reagent, timing, transfection volume and temperature. Tools such as Design of Experiments (DoE) can be particularly useful in multifactorial experiments such as these. Compared to the experimental approach of changing one factor at a time, DoE allows one to examine more than one factor in an experiment and identify interactions between factors affecting a process and its outputs. Furthermore, optimization of these parameters can help to establish robustness that can be used to address limitations related to scale-up of the transient transfection cocktail (i.e., mixing, timing, addition, etc.). Still, development scientists should consider the quadratic effects of other process variables such as pH or DO that cannot be sufficiently maintained in shake-flask studies through scaled demonstration runs. Prior to proceeding with cGMP production, it is often a good idea to perform engineering runs at full production scale. Although these runs are an added cost, it is possible to utilize this material for toxicology/biodistribution studies, reference standards, comparability studies, as well as for in-process and product quality/analytical characterization.
Lastly, single-use stirred-tank bioreactors have become one of the most commonly used reactor types in the industry. Designed based on traditional stainless-steel reactor mixing principles, they offer benefits such as lower start-up and implementation costs as well as reduced clean-in-place and steam-in-place (CIP/SIP). Most commercially available single-use bioreactor platforms are engineered to meet the needs of many mammalian cell culture processes. That said, caution should be exercised when selecting a platform based on compatibility of cell types, media components and therapeutic products to the plastics/films used in the material of construction as well as the inherent scalability offered by the manufacturer.
Purification
Downstream unit operations used in viral vector manufacturing are similar to those of other biomolecules. One of the critical roadblocks that the industry is facing at the moment is that, unlike other bioprocesses, such as mAbs, gene therapy manufacturing has not experienced decades of optimization and characterization yielding high titers that have influenced the scale of equipment currently available on the market today. For this reason, it’s often difficult to work out reliable models for both process development and early stage clinical production.
Although there have been substantial improvements in the upstream scale of rAAV production, there is a relatively low concentration of target drug product in the crude harvest of most manufacturing platforms when compared to other biologics. As upstream productivities increase, downstream processes need to adapt with purification strategies that maintain rAAV vector yields that exhibit good safety and efficacy, as well as high purity profiles. To date, many reported purification methods are serotype dependent, requiring capsid-specific process development for each rAAV gene therapy product [36,37]. While they may have been originally developed with the intent of being a universal platform for purification, manufacturers often result to serotype-specific optimizations as the characteristics of the vectors can be variable. This is observed even in the case of many commercially available affinity resins that are advertised as universal, but still display variable chromatographic profiles with respect to the natural serotypes [38]. Nass et al. recently reported a method compatible with multiple AAV serotypes produced in the transient triple transfection system and utilizing a two-column purification method, based on affinity and ion exchange chromatographies [39]. Nonetheless, it is expected that some process changes will need to be incorporated when transitioning from early phase to late phase clinical stages. For example, the use of ultracentrifugation [6,40] is the most common method utilized to enrich for full capsids, yet it is probably the most commonly recognized bottleneck as it is difficult to scale to commercial manufacturing.
One hurdle to overcome in most rAAV production platforms is the production of empty particles, or viral capsids that do not contain a full-length therapeutic vector genome. Viewed as a product-related impurity, empty particles are a concern as they may elicit unwanted host immune responses in both humans and animals due to the increased exposure to AAV antigen [41–43]; however, this perspective remains controversial [44,45]. There have been claims that improvements have been made to upstream production systems that control the amount of full particles in the harvest, however, this data remains to be reported. Without comparative studies across production platforms using improved analytical assays to accurately quantify empty and full particles such as analytical ultracentrifugation (AUC) [46] or cryo-electron microscopy (cryo-EM) [47] it is difficult to claim that one is superior to the other. Still, the problem with scaling up process steps such as gradient ultracentrifugation remains. On the other hand, there have been downstream processing methods described that support the chromatographic separation of empty and full particles by traditional ion-exchange chromatography based on slight charge differences between the two [48,49].
QC release & regulatory perspectives
A robust manufacturing process is essential to ensure the correct quality and safety of a biological product. Yet because of their larger size, complex structure, formulation, and manufacture, rAAV vectors can pose a challenge in developing comprehensive analytical assays for product characterization. There is an apparent need for manufacturers to invest in improving the analytical techniques currently being used to quantify and characterize rAAV vectors. For example, development of improved titering methods and in vitro potency assays are critical for the advancement of rAAV gene therapies. In the case of the vector genome titer, methods have evolved from the DNA dot-blot to quantitative PCR (qPCR) to now digital droplet PCR (ddPCR), but efforts should still be focused on reducing variation, improving sensitivity, precision and accuracy to fully understand and define critical quality attributes (CQAs) that impact product safety, purity and potency.
In general, manufacturing experience with any given rAAV vector is limited due to varying AAV serotype, vector transgene, as well as dose requirements. Therefore, any early stage clinical candidate, such as material from academic production facilities, will not have sufficient historical analytical information to allow for final specifications to be established for various quality attributes or predict the impact of changes in quality attributes. It is important in early stage product development that analytical data be collected using qualified assays to demonstrate batch-to-batch consistency, allow for tighter specifications to be set, and establish fully validated assays after sufficient manufacturing experience with a particular vector has been gained. Product testing for safety attributes such as sterility, mycoplasma, adventitious agents (replication-competent AAV) must be qualified and validated, with the overall testing plan in place to ensure that the product meets acceptable limits for identity, strength/potency, quality, and purity as noted in the FDA’s Guidance for Industry in: July 2008, cGMP for Phase 1 Investigational Drugs; and April 2008, Content and Review CMC Information for Human Gene Therapy IND Applications. Lastly, as it is likely that process changes will be implemented through optimization and scale up efforts, it is essential to ensure that sample retains (i.e., drug substance, in-process samples, Ph I clinical drug product) are collected for analytical comparability exercises.
Conclusion
With promising clinical data as well as continued success in pre-clinical studies, rAAV-mediated gene therapies have risen to the threshold of a new and exciting class of molecular medicine. Yet manufacturing sufficient quantities of vector to meet the rapidly growing demand for clinical product still poses significant challenges on the path to late stage clinical trials and commercialization. Although many advances have been made to all rAAV production platforms, manufacturing technologies as well as many of the supporting analytical assays, are still in their infancy when compared to standard biologicals. Several of the bottlenecks facing the scale up and commercialization of rAAV gene therapy products will require a concerted effort not only between academic and industrial drug developers, but also with equipment and raw material manufacturers to address limitations in rAAV production technologies, supporting analytical assays, critical equipment and raw materials. For efficient transition between early stage clinical trials and commercialization, it is critical that investments are made early in the development of manufacturing processes and analytical characterization to meet the increased rAAV vector needs and regulatory demands especially if transitioning to a CMO.
Financial & competing interests disclosure
JMS is an employee of Asklepios Biopharmaceutical, Inc. JCG is an employee of Asklepios Biopharmaceutical, Inc. and holds patents that have been licensed by the University of North Carolina to Asklepios Biopharmaceutical, Inc for which he receives royalties. RJS is a founder and shareholder of Asklepios Biopharmaceutical, Inc. He holds patents that have been licensed by the University of North Carolina to Asklepios Biopharmaceutical, Inc for which he receives royalties.
No writing assistance was utilized in the production of this manuscript.
References
1. Srivastava A, Lusby EW, Berns KI. Nucleotide sequence and organization of the adeno-associated virus 2 genome. J. Virol. 1983; 45(2): 555–64. CrossRef
2. Sonntag, F., et al., The assembly-activating protein promotes capsid assembly of different adeno-associated virus serotypes. J. Virol. 2011; 85(23): 12686-97. CrossRef
3. Samulski RJ, Köther K, Schmidt K et al. Cloning of adeno-associated virus into pBR322: rescue of intact virus from the recombinant plasmid in human cells. Proc. Natl Acad. Sci. USA 1982; 79(6): 2077–81. CrossRef
4. Hermonat PL, Muzyczka N. Use of adeno-associated virus as a mammalian DNA cloning vector: transduction of neomycin resistance into mammalian tissue culture cells. Proc. Natl Acad. Sci. USA 1984; 81(20): 6466–70. CrossRef
5. Xiao X., Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 1998; 72(3): 2224–32.
6. Grieger JC, Soltys SM, Samulski RJ. Production of Recombinant Adeno-associated Virus Vectors Using Suspension HEK293 Cells and Continuous Harvest of Vector From the Culture Media for GMP FIX and FLT1 Clinical Vector. Mol. Ther. 2016; 24(2): 287–97. CrossRef
7. Martin J, Frederick A, Luo Y et al. Generation and characterization of adeno-associated virus producer cell lines for research and preclinical vector production. Hum. Gene Ther. Methods 2013; 24(4): 253–69. CrossRef
8. Martin JN, Wolken N, Brown T, Dauer WT, Ehrlich ME, Gonzalez-Alegre P. Lethal toxicity caused by expression of shRNA in the mouse striatum: implications for therapeutic design. Gene Ther. 2011; 18(7): 666–73. CrossRef
9. Liu XL, Clark KR, Johnson PR. Production of recombinant adeno-associated virus vectors using a packaging cell line and a hybrid recombinant adenovirus. Gene Ther. 1999; 6(2): 293–9. CrossRef
10. Galibert L, Merte OW. Latest developments in the large-scale production of adeno-associated virus vectors in insect cells toward the treatment of neuromuscular diseases. J. Invertebr. Pathol. 2011; 107 Suppl: S80–93. CrossRef
11. Urabe M, Ding C, Kotin RM. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum. Gene Ther. 2002; 13(16): 1935–43. CrossRef
12. Smith RH, Levy JR, Kotin RM. A simplified baculovirus-AAV expression vector system coupled with one-step affinity purification yields high-titer rAAV stocks from insect cells. Mol. Ther. 2009; 17(11): 1888–96. CrossRef
13. Mietzsch M, Grasse S, Zurawski C et al. OneBac: platform for scalable and high-titer production of adeno-associated virus serotype 1-12 vectors for gene therapy. Hum. Gene Ther. 2014; 25(3): 212–22. CrossRef
14. Mietzsch M, Casteleyn V, Weger S et al. OneBac 2.0: Sf9 Cell Lines for Production of AAV5 Vectors with Enhanced Infectivity and Minimal Encapsidation of Foreign DNA. Hum. Gene Ther. 2015; 26(10): 688–97. CrossRef
15. Cecchini S, Virag T, Kotin RM. Reproducible high yields of recombinant adeno-associated virus produced using invertebrate cells in 0.02- to 200-liter cultures. Hum. Gene Ther. 2011; 22(8): 1021–30. CrossRef
16. Aucoin MG, Mena JA, Kamen AA. Bioprocessing of baculovirus vectors: a review. Curr. Gene Ther. 2010; 10(3): 174–86. CrossRef
17. Adamson-Small L, Potter M, Falk DJ, Cleaver B, Byrne BJ, Clement, N. A scalable method for the production of high-titer and high-quality adeno-associated type 9 vectors using the HSV platform. Mol. Ther. Methods Clin. Dev. 2016; 3: 16031. CrossRef
18. Clement N, Knop DR, Byrne BJ. Large-scale adeno-associated viral vector production using a herpesvirus-based system enables manufacturing for clinical studies. Hum. Gene Ther. 2009; 20(8): 796–06. CrossRef
19. Penaud-Budloo M, François A, Clément N, Ayuso E. Pharmacology of Recombinant Adeno-associated Virus Production. Mol. Ther. Methods Clin. Dev. 2018. 8: p. 166-180. CrossRef
20. Grimm D, Kern A, Rittner K, Kleinschmidt JA. Novel tools for production and purification of recombinant adeno-associated virus vectors. Hum Gene Ther. 1998; 10; 9(18): 2745-60.
21. Graham, F. L., Smiley, J., Russell, W. C., & Nairn, R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977; 36(1): 59–74. CrossRef
22. Weindler FW, Heilbronn R. A subset of herpes simplex virus replication genes provides helper functions for productive adeno-associated virus replication. J Virol. 1991; 65(5): 2476-83.
23. Solid Biosciences Announces Clinical Hold On SGT-001 Phase I/II Clinical Trial For Duchenne Muscular Dystrophy, 2018. https://investors.solidbio.com/news-releases/news-release-details/solid-biosciences-announces-clinical-hold-sgt-001-phase-iii
24. Schnodt M, Schmeer M, Kracher B et al. DNA Minicircle Technology Improves Purity of Adeno-associated Viral Vector Preparations. Mol. Ther. Nucleic Acids 2016; 5: e355. CrossRef
25. Schnodt M, Buning H. Improving the Quality of Adeno-Associated Viral Vector Preparations: The Challenge of Product-Related Impurities. Hum. Gene Ther. Methods 2017; 28(3): 101–8. CrossRef
26. Karbowniczek K, Rothwell P, Extance J et al. DoggyboneTM DNA: an advanced platform for AAV production. Cell Gene Ther. Insights 2017; 3(9): 731–8. CrossRef
27. European Medicines Agency (EMA). 2012 Assessment report: Glybera: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002145/WC500135476.pdf.
28. Merten O, Schweizer M, Chahal P, Kamen AA. Manufacturing of viral vectors for gene therapy: part I, Upstream processing. Pharm. Bioprocess 2014; 2( 2): 183–203. CrossRef
29. Pfizer Aims To Become Industry Leader In Gene Therapy With Acquisition Of Bamboo Therapeutics, Inc., 2016: https://www.pfizer.com/news/press-release/press-release-detail/pfizer_aims_to_become_industry_leader_in_gene_therapy_with_aquisition_of_bamboo_therapeutics_inc
30. Exclusive technology for large-scale cGMP manufacturing of AAV vectors, 2017: http://www.viralgenvc.com/technology/
31. Baxter to Acquire Chatham Therapeutics, 2014: https://investor.baxter.com/investors/events-and-news/news/press-release-details/2014/Baxter-to-Acquire-Chatham-Therapeutics/default.aspx
32. FDA Places Hold on Phase 1/2 Trial of Sarepta’s Gene Therapy for DMD, 2018: https://musculardystrophynews.com/2018/07/30/fda-places-clinical-hold-trial-sarepta-duchenne-gene-therapy
33. Kaczmarek R. Do adventitious viruses carried by insect cell lines producing AAV vectors pose a safety risk in gene therapy? Haemophilia 2018; doi:10.1111/hae.13525 CrossRef
34. Ma H, Galvin TA, Glasner DR, Shaheduzzaman S, Khan AS. Identification of a novel rhabdovirus in Spodoptera frugiperda cell lines. J. Virol. 2014; 88(12): 6576–85. CrossRef
35. Snyder, R., Session 4: Quality and Manufacturing. The Growing Promise of Gene Therapy Approaches to Rare Diseases workshop, 2018. Bethesda, MD, USA.
36. O’Riordan CR, Lachapelle AL, Vincent KA et al. Scaleable chromatographic purification process for recombinant adeno-associated virus (rAAV). J. Gene Med. 2000; 2(6): 444–54. CrossRef
37. Zhou J, Yang X, Wright JF et al. PEG-modulated column chromatography for purification of recombinant adeno-associated virus serotype 9. J. Virol. Methods 2011; 173(1): 99–107. CrossRef
38. Wang Q, Lock M, Prongay AJ et al. Identification of an adeno-associated virus binding epitope for AVB sepharose affinity resin. Mol. Ther. Methods Clin. Dev. 2015; 2: 15040. CrossRef
39. Nass SA, Mattingly MA, Woodcock DA et al. Universal Method for the Purification of Recombinant AAV Vectors of Differing Serotypes. Mol. Ther. Methods Clin. Dev. 2018; 9: 33–46. CrossRef
40. Wright JF, Wellman J, Hig KA. Manufacturing and regulatory strategies for clinical AAV2-hRPE65. Curr. Gene Ther. 2010; 10(5): 341–9. CrossRef
41. Manno CS, Pierce GF, Arruda VR et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006; 12(3): 342–7. CrossRef
42. Gao K, Li M, Zhong L, Su Q, Li J, Li S, Gao G. Empty Virions In AAV8 Vector Preparations Reduce Transduction Efficiency And May Cause Total Viral Particle Dose-Limiting Side-Effects. Mol. Ther. Methods Clin. Dev. 2014; 1(9): 20139. CrossRef
43. Pei X, Earley LF, He Y, Chen X, Hall NE, Samulski RJ, Li C. Efficient Capsid Antigen Presentation From Adeno-Associated Virus Empty Virions In Vivo. Front. Immunol. 2018; 9: 844. CrossRef
44. Aalbers CJ, Broekstra N, van Geldorp M et al. Empty Capsids and Macrophage Inhibition/Depletion Increase rAAV Transgene Expression in Joints of Both Healthy and Arthritic Mice. Hum. Gene Ther. 2017; 28(2): 168–78. CrossRef
45. Mingozzi F, Anguela XM, Pavani G et al. Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci. Transl. Med. 2013; 5(194): 194ra192. CrossRef
46. Burnham B, Nass S, Kong E et al. Analytical Ultracentrifugation as an Approach to Characterize Recombinant Adeno-Associated Viral Vectors. Hum. Gene Ther. Methods 2015; 26(6): 228–42. CrossRef
47. Full or empty analysis, 2018: https://www.vironova.com/analyzes/full-empty/
48. Qu G, Bahr-Davidson J, Prado J et al. Separation of adeno-associated virus type 2 empty particles from genome containing vectors by anion-exchange column chromatography. J. Virol. Methods 2007; 140(1-2): 183–92. CrossRef
49. Lock M, Alvira MR, Wilson JM. Analysis of particle content of recombinant adeno-associated virus serotype 8 vectors by ion-exchange chromatography. Hum. Gene Ther. Methods 2012; 23(1): 56–64. CrossRef
Affiliations
Jacob M Smith1, Joshua C Grieger1, R Jude Samulski2
1 Asklepios Biopharmaceutical Inc., Durham, North Carolina, USA
Gene Therapy Center, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.