Doggybone™ DNA: an advanced platform for AAV production
Cell Gene Therapy Insights 2017; 3(9), 731-738.
10.18609/cgti.2017.074
Recombinant adeno-associated virus (AAV) represents one of the most promising delivery vehicles for genetic medicines. However, the manufacture of plasmid DNA for the production of AAV presents a number of significant challenges, including scalability, fidelity, mis-incorporation of plasmid-derived DNA sequences, high costs and long lead times for GMP production. Touchlight has developed a novel, rapid, in vitro, enzymatic technology for multi-gram scale GMP manufacture of DNA that addresses all of the issues of DNA manufacture for AAV production. The process combines the use of two enzymes; Phi29 DNA polymerase and a protelomerase to generate covalently closed, linear DNA constructs known as doggybone™ DNA or dbDNA™. The process is rapid, cost effective, of high fidelity and eliminates antibiotic resistance genes. Here we present a case study, with data generated by Cobra Biologics as part of an on-going Innovate UK-funded collaboration, demonstrating that AAV particles can be produced using dbDNA™ with total and genomic titres equivalent to AAV particles made with plasmid DNA. In parallel experiments, with an academic collaborator, we have seen in vivo expression of reporter genes after administration of AAV vectors manufactured using dbDNA™ (unpublished data). As such, we believe that dbDNA™ has the potential to resolve a number of significant challenges in the production of AAV vectors at both clinical and commercial scale.
Recombinant adeno-associated virus (AAV) is broadly considered to be the most promising delivery vehicle for genetic medicines, and such treatments have the potential to address a wide range of previously intractable genetic conditions [1]. At present, hundreds of AAV products are being assessed in early- and late-stage clinical trials [2], with the first Biologics Licensing Application to the FDA made by Spark Therapeutics in May 2017. Multiple systems for the manufacture of AAV have been developed but currently the co-transfection of HEK293 cells, typically using three bacterial plasmids, is the most widely used. In this method, plasmids encoding non-structural, structural and helper elements are provided in trans. Rep and cap elements encode viral replication and capsid proteins, respectively, and a helper plasmid encodes a number of adenovirus gene products required for efficient AAV replication. A third viral transgene plasmid contains a genetic payload flanked by cis-acting inverted terminal repeats (ITRs) necessary for transgene rescue and packaging [1,3].
Whilst transient transfection is currently considered the gold standard for clinical and commercial manufacture of AAV, the production and use of plasmid DNA presents a number of challenges and introduces a significant bottleneck for agile discovery and commercial-scale manufacture. First, the cruciform secondary structures of the cis-acting ITR sequences can be difficult to propagate in Escherichia coli, and plasmid preparations frequently contain deletions in the ITR regions [4]. Second, plasmid DNA production typically requires antibiotic selection, resulting in transfected plasmids containing backbone sequences encoding antibiotic resistance genes. Plasmid-derived sequences are known to be packaged into AAV capsids at a frequency estimated at 1–5% [5] but packaging has been reported to be as high as 26.1% [6]. Finally, the majority of production cost of plasmid DNA in E. coli comprises time in GMP suite, capital and labor costs, which represents a challenge for capacity expansion of the existing market. With current timelines for CMO production of gram scale GMP DNA production at several months [7], the rapidly advancing gene therapy pipeline is experiencing a significant bottleneck in DNA supply.
Here we describe a novel, rapid, in vitro, enzymatic technology for multi-gram scale GMP manufacture of DNA for AAV production. The process combines the use of two enzymes – Phi29 DNA polymerase and a protelomerase, and generates high fidelity, covalently closed, linear DNA constructs known as doggybone™ DNA or dbDNA™. dbDNA™ constructs contain no antibiotic resistance markers, and therefore eliminate the packaging of these sequences. Our results demonstrate that the process can amplify AAV constructs in a 2-week process at commercial scale and maintain the ITR sequences required for virus production. As demonstrated in the case study described here, when evaluated by Cobra Biologics using their AAV production approach, dbDNA™ can be used to produce functional AAV particles, and can serve as a complete replacement for plasmid DNA. Additional studies [Unpublished Data] have established a robust, efficient platform for AAV production using dbDNA™. We anticipate that this technology will resolve a number of significant issues in the production of AAV vectors at both clinical and commercial scale.
The use of minimal constructs that remove antibiotic resistance genes, such as minicircle DNA, in addition to the use of linear, covalently-closed DNA structures, such as ‘no-end’ DNA have been reported for the small scale production of AAV [6,8,9]. However, pDNA has remained the gold standard – most likely due to the inability to scale these previous technologies for commercial use. A technology for the manufacture of a minimal construct, such as dbDNA™, at commercial scale and quality could offer a viable alternative system for AAV production.
Results
Overview of dbDNA™ technology
We have developed a purely in vitro DNA amplification and purification technology for the production of DNA at large scale (Figure 1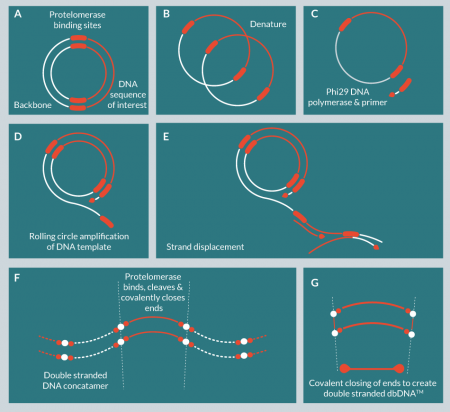
Phi29 DNA polymerase was selected for this process because of its high fidelity (1/106–1/107) and high processivity (approximately 70 kbp) [10]. These features made this polymerase particularly suitable for the large-scale production of GMP DNA. Protelomerases (also known as telomere resolvases) catalyze the formation of covalently closed hairpin ends on linear DNA and have been identified in some phages, bacterial plasmids and bacterial chromosomes [11–13]. A pair of protelomerases recognizes inverted palindromic DNA recognition sequences and catalyzes strand breakage, strand exchange and DNA ligation to generate closed linear hairpin ends [14]. The formation of these closed ended structures makes the DNA resistant to exonuclease activity, allowing for simple purification and can improve stability and duration of expression [15].
The production of dbDNA™ depends upon the introduction of protelomerase recognition sequences that flank the gene of interest. These sites are 56 bp palindromic sequences that are highly specific to particular protelomerase enzymes (Figure 2
These constructs are termed doggybone™ DNA, or dbDNA™, as a reference to the shape of the construct when the closed ends are schematically exaggerated in cartoon form (Figure 2). dbDNA™ molecules are minimal constructs, containing only a sequence of interest flanked by half-protelomerase binding sequences resulting from concatamer resolution. When denatured, dbDNA™ constructs comprise circular DNA molecules that can be used as a starting material for further amplification reactions.
Development of a scalable, GMP, benchtop process for dbDNA™ production
We have developed a manufacturing process in accordance with EU GMP ICH Q7. dbDNA™ can currently be produced as a critical starting material for the GMP manufacture of viral vectors. Additionally, the production process is compatible with the production of dbDNA™ as an active pharmaceutical ingredient (API). The process takes 2 weeks, including QC, and is performed by a single operator in a 10m2 production pod. dbDNA™ production is performed entirely with disposable contact materials and with low capital cost laboratory equipment, including benchtop centrifuges and static incubators.
The process has been demonstrated to be linearly scalable to this point (Figure 3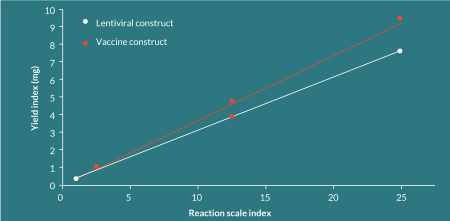
The cost composition of this completely abiological process contrasts significantly with that for the manufacture of plasmid DNA. The process has minimal operator time, a short duration (1 week in the manufacturing facility), low-cost equipment and a benchtop footprint, leading to low fixed costs that are largely independent of batch size, providing significant economies of scale. Because the process is abiological, the requirement for large, dedicated facilities with high capital requirements is removed. As a result, in excess of 90% of the cost of a batch of dbDNA™ comprises of the cost of reagents and variable material costs.
AAV sequence production using dbDNA™
As part of an Innovate UK-funded collaboration between Touchlight and Cobra Biologics, we performed a case study to evaluate the performance of dbDNA™ for the production of AAV by transient transfection. Key questions included: 1) whether the technology is able to amplify the relevant sequences without rearrangements or loss of yield; 2) whether the fidelity of the ITR sequences is maintained in the resulting transgene construct; and 3) whether triple transfection using only dbDNA™ produces functional AAV.
To evaluate dbDNA™ for production of viable AAV particles, we designed three dbDNA™ sequences; Helper, RepCap and a Payload encoding an eGFP reporter. Target dbDNA™ constructs were identical to their plasmid DNA counterparts in terms of the components required to produce functional AAV but excluded the extraneous backbone sequences required for plasmid DNA propagation in E. coli.
We successfully amplified all three dbDNA™ constructs at high yield and each was purified using our standard method. Figure 4 (lane 2) depicts the result of the amplification and purification of a dbDNA™ Payload construct, showing the product as a single species on agarose gel electrophoresis.
Additionally, restriction enzyme digests targeting cut sites within the ITRs confirmed that both ITR sequences were maintained during amplification of the payload construct (Figure 4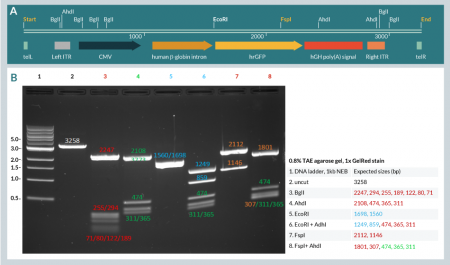
Production of virus from dbDNA
To assess the ability of dbDNA™ to generate AAV particles, linear PEI:DNA complexes (1:1 PEI:DNA) were transfected into HEK293T suspension adapted cells bound to CellBIND plates at a molar ratio previously optimized by Cobra Biologics, and in parallel to plasmid DNA transfections. A transient transfection protocol, previously developed by Cobra Biologics, was performed on HEK293 cells with the Helper, RepCap and Payload. AAV particles were released from the cells by detergent lysis. Genomic particle titres were determined using ITR q-PCR and total particle titres using an AAV2 capsid ELISA (Figure 5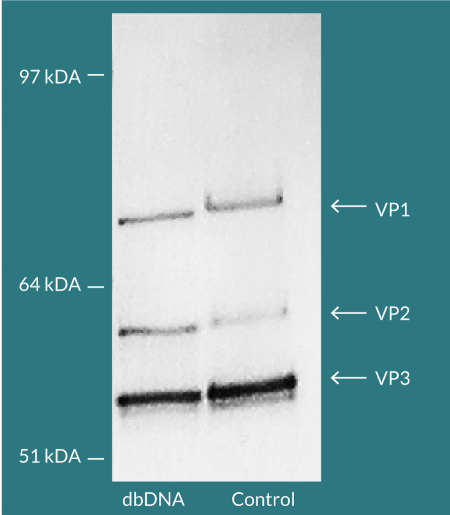
Comparable total and genomic titers were achieved for pDNA and dbDNA™- produced AAV confirming the substitutability of using dbDNA™ in AAV manufacture. Viral capsid protein ratios that closely matched an AAV reference sample were evidenced by Western blot (Figure 6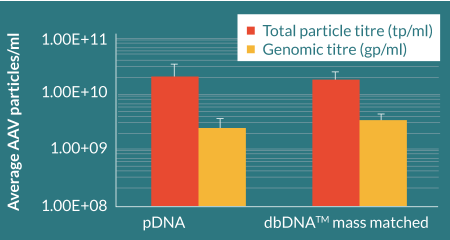
Discussion
With the anticipated success of AAV gene therapy, the ability to manufacture large quantities of DNA at GMP quality is critical to scaling these products to meet demand. Traditional methods of pDNA production are time consuming and costly due to the need for large-scale fermentation and downstream processing to make gram quantities of GMP DNA. AAV production delays due to a bottleneck in pDNA supply could be highly deleterious to discovery and clinical development efforts.
The technology to produce dbDNA™ is able to meet these needs. The enzymatic dbDNA™ technology has been shown to be scalable, rapid, amenable to GMP production and is low cost. The case study shown here demonstrates that the technology can amplify and maintain ITR-containing AAV payload sequences, and that when combined and optimized within existing platforms, triple-dbDNA™ transfection produces functional AAV at titers at least comparable to those produced with an equivalent plasmid DNA system. Additional studies have been performed at larger scale and with further purification that further confirm the utility and benefits of dbDNA™ in the AAV system [Manuscript in Preparation]. In conclusion, the novel technology developed for dbDNA™ production represents an important advance that can enable the success of AAV gene therapy.
financial & competing interests disclosure
KK, PR, JE, SM & LC are employees of Touchlight Genetics; VL, KB & DS are employees of Cobra Biologics. They have no financial conflict with the subject matter or materials discussed in the manuscript. No writing assistance was utilized in the production of this manuscript.
References
1. Samulski RJ, Muzyczka N. AAV-Mediated Gene Therapy for Research and Therapeutic Purposes. Annu. Rev. Virol. 2014; 1: 427–51.
CrossRef
2. Pharma eTrack database, GlobalData (pull: May 2017).
3. Kotin RM. Large-scale recombinant adeno-associated virus production. Hum. Mol. Genet. 2011; 20(R1): R2–6.
CrossRef
4. Yan Z, Zak R, Zhang Y, Engelhardt JF. Inverted Terminal Repeat Sequences Are Important for Intermolecular Recombination and Circularization of Adeno-Associated Virus Genomes. J. Virol. 2005; 79(1): 364–79.
CrossRef
5. Chadeuf G, Ciron C, Moullier P, Salvetti A. Evidence for encapsidation of prokaryotic sequences during recombinant adeno-associated virus production and their in vivo persistence after vector delivery. Mol. Ther. 2005; 12(4): 744–53.
CrossRef
6. Schnödt M, Schmeer M, Kracher B et al. DNA Minicircle Technology Improves Purity of Adeno-associated Viral Vector Preparations. Mol. Ther. Nucleic Acids 2016; 5(8): e355.
CrossRef
7. VGXI, Inc. 2012. How Long Does It Take to Manufacture Plasmid under GMP? White paper: Website
8. Nahreinia P, Larsenb SH, Srivastava A. Cloning and integration of DNA fragments in human cells via the inverted terminal repeats of the adeno-associated virus 2 genome. Gene 1992; 119: 265–72.
CrossRef
9. Snyder RO, Im DS, Muzyczka N. Evidence for covalent attachment of the adeno-associated virus (AAV) rep protein to the ends of the AAV genome. J. Virol. 1990; 12: 6204–13.
10. Garmendia C, Bernad A, Esteban JA, Blanco L, Salas M. The bacteriophage phi 29 DNA polymerase, a proofreading enzyme. J. Biol. Chem. 1992; 267(4): 2594–9.
11. Deneke J, Ziegelin G, Lurz R, Lanka E. The protelomerase of temperate Escherichia coli phage N15 has cleaving-joining activity. Proc. Natl Acad. Sci. USA 2000; 97: 7721–6.
CrossRef
12. Kobryn K, Chaconas G. ResT, a telomere resolvase encoded by the Lyme disease spirochete. Mol. Cell 2002; 9: 195–201.
CrossRef
13. Huang WM, DaGloria J, Fox H et al. Linear chromosome-generating system of Agrobacterium tumefaciens C58: protelomerase generates and protects hairpin ends. J. Biol. Chem. 2012; 287: 25551–63.
CrossRef
14. Huang WM, Joss L, Hsieh TT, Casjens S. Protelomerase uses a topoisomerase IB/Y-recombinase type mechanism to generate DNA hairpin ends. J. Mol. Biol. 2004; 337: 77–92.
CrossRef
15. Lina Li, Dimitriadis EK, Yang Y et al. Production and Characterization of Novel Recombinant Adeno-Associated Virus Replicative-Form Genomes: A Eukaryotic Source of DNA for Gene Transfer. PLoS ONE 2013; 8(8): e69879.
CrossRef
Affiliations
Kinga Karbowniczek1, Paul Rothwell1, Jon Extance1, Sarah Milsom1, Vera Lukashchuk2, Kevin Bowes2, Daniel Smith2 & Lisa Caproni1
1 Touchlight Genetics Ltd
2 Cobra Biologics Ltd

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.