Toward an integrated model of product characterization for CAR-T cell therapy drug development efforts
Cell Gene Therapy Insights 2017; 3(4), 227-237.
10.18609/cgti.2017.026
Chimeric antigen receptor T (CAR-T) cell therapies have produced remarkable clinical outcomes in relapse/refractory cancer patients. These therapies have also led, in certain cases, to life-threatening side effects. The field is now at a critical juncture where a number of CAR-T cell therapies have completed pivotal trials and are on the path to global registration. However, little is known about how to characterize these ‘living’ drug products before administration and more needs to be understood about how different manufacturing strategies can affect safety and efficacy. Moreover, the interplay between the CAR-T cell therapy drug product and the host-specific microenvironment remains incompletely understood. This brief article will review some of the most common analytical challenges encountered during CAR-T cell therapy drug product development and will propose an integrated analytical characterization strategy that may enable for the rapid and comprehensive development of safer and more effective CAR-T therapies.
CAR-T cell therapies in the Clinic
Chimeric antigen receptors (CARs) are synthetic, engineered receptors that can target surface molecules in their native conformation [1]. Unlike T cell receptors (TCRs), CARs engage molecular structures independent of antigen processing by the target cell, and independent of major histocompatibility complex [1]. CARs typically engage the target via a single-chain variable fragment (scFv) derived from an antibody, although natural ligands have also been used [2]. Individual scFvs targeting a surface molecule are either derived from murine or humanized antibodies or synthesized and screened via phage display libraries [3]. CAR-T therapies are living therapies, which consist of the genetic reprogramming of a patient’s own T cells to express CARs directed against targets expressed by malignant cells [1-3].
The most dramatic and consistent clinical outcomes to date, have been obtained with CD19-directed CAR-T cell therapies in patients with relapse/refractory B-cell derived hematologic malignancies; adults and children with acute lymphocytic leukemia (ALL) have shown the most durable complete response (CR) rates (Table 1). Despite the extraordinary success of CD19 CAR-T cell therapies, the specific active ingredient(s) of the drug product are not well understood. There are multiple factors that likely contribute to CAR-T drug product activity including: optimal vector and transduction method, culture conditions, CAR design, T-cell type, and dose of the cell type. Moreover, there are a complex set of tumor, host and environmental factors that govern the strength and timing of the anticancer response mediated by the infused CAR-T cell therapy product [4].
| Table 1: CD19 CAR-T cell therapies in the clinic. | ||||||
|---|---|---|---|---|---|---|
| Center | Treated patients (n) | Disease | Potency Assay | Objective responder | Non-responder | Ref. |
| City of Hope | 2 | Lymphoma | 51Cr release | 0 | 2 | [26] |
| Baylor | 6 | Lymphoma | 51Cr release | 0 | 6 | [27] |
| NCI | 8 | 4 CLL 4 Lymphoma | IFN- γ release | 6 | 2 | [28,29] |
| MSKCC | 9 | 8 CLL 2 ALL | 51Cr release | 1 | 8 | [20] |
| Upenn | 3 | CLL | IFN- γ | 1 2 | 0 0 | [18,19] |
| MSKCC | 16 | ALL | 51Cr release | 2 12 | 2 0 | [30,31] |
| NIH/NCI | 10 | 4 CLL 6 Lymphoma | IFN- γ | 2 | 8 | [32] |
| NIH/NCI | 21 | 20 ALL 1 Lymphoma | ? | 14 | 7 | [33] |
| UPenn | 30 | ALL | IFN- γ | 25 | 5 | [25,34] |
| Kite ZUMA-1 | 101 | Lymphoma | IFN- γ | 41 | 60 | [35] |
| Novartis CTL019 | 50 | ALL | IFN- γ | 41 | 9 | [36] |
| The table shows a subset of the CD19 directed CAR-T cell therapy trials for relapse/refractory B cell derived hematological malignancies that have been conducted to date. The table also includes a listing of the potency assay, when available, used for product release for clinical use of the CAR-T cell product. It should be noted that the UPenn and CTL019 clinical trials for ALL that are cited in this table used the same overall CAR-T cell therapy manufacturing process and clinical trial design. | ||||||
CAR-T cell therapies also have the potential to trigger, in some cases, life threatening severe adverse events including cytokine release syndrome, tumor lysis syndrome, B-cell aplasia and neurological toxicity (Table 2). The toxic potential of CAR-T cell therapies was most strikingly revealed in the Juno Therapeutics sponsored JCAR015 program for relapse or refractory adult B-cell ALL patients. In July 2016, three patients experienced fatal cerebral edema following infusion with Juno Therapeutics’ CD19 CAR-T cell therapy, which is a retrovirus based mixed T cell product [5]. Juno Therapeutics speculated that the fatalities were due to the combined cyclophosphamide and fludarabine pre-conditioning regimen [5]. After a brief clinical trial hold, Juno Therapeutics resumed the ROCKET trial completely eliminating fludarabine from the pre-conditioning regimen. Despite this less intensive regimen, two additional patients experienced fatal cerebral edema on the ROCKET trial in November 2016. After the second set of patient deaths, Juno concluded that the tragic results reflected “patient-specific factors, the conditioning chemotherapy patients received, and factors related to the product” [6]. In essence, both drug product-specific and patient-specific factors led to the unexpected toxicities.
The CD19 CAR-T cell therapy clinical experience, with respect to efficacy (Table 1) and safety (Table 2), highlights the need for organizations to create a unified analytical characterization strategy that transcends the traditional boundaries of drug development efforts. This brief article will highlight some of the major challenges and opportunities for CAR-T cell therapy drug product development.
| Table 2: Clinical adverse events associatedwith CD19 CAR-T cell therapy. |
|---|
| Cytokine release syndrome (CRS) [25,37–39] |
|
| Tumor lysis syndrome (TLS) [40–42] |
|
| Sustained B cell aplasia [42,43] |
|
| Neurological toxicity [43,44] |
|
| The table discusses the most common clinical adverseevents associatedwithCD19 CAR-T cell therapies. |
Analytical challenges related to product characterization
CAR-T cell therapy manufacturing requires several carefully performed steps including: lymphocyte collection and enrichment, lymphocyte activation, genetic modification with the CAR construct, lymphocyte expansion, harvest and final formulation (Figure 1).
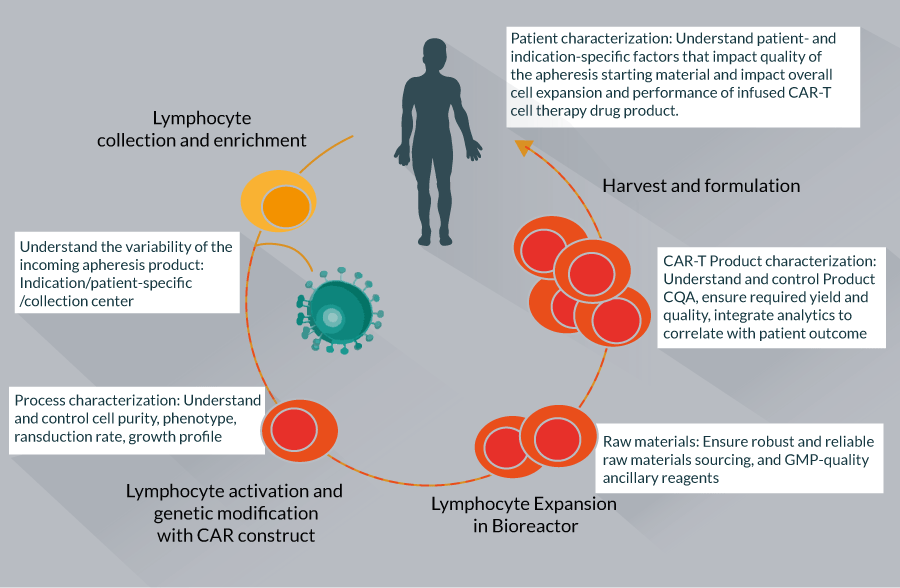
Figure 1 The figure highlights the common steps that underpin the majority of CAR-T cell therapy manufacturing eff orts and highlights the
overall aim of analytical characterization for each step. As can be seen, the CAR-T manufacturing process blurs traditional drug development line functions.
The manufacturing process entails the use of reagents and source materials of differing complexity, variability and risk for introduction of adventitious agents. Qualification of reagents and source materials, as well as ensuring that appropriate controls are in place for monitoring manufacturing consistency and product quality, are all key elements in ensuring that subjects receive a product that is consistently safe, pure, and potent. Within this manufacturing framework, identifying and monitoring critical quality attributes (CQAs) that associate with manufacturing and clinical success becomes the primary objective of the overall analytical characterization strategy for manufacturing of CAR-T cell therapies. In a nutshell, CQAs are chemical, physical, biological and microbiological attributes that can be defined, measured and continually monitored to ensure final product outputs remain within acceptable quality limits. CQAs are the manufacturing equivalent of biomarkers. Whereas biomarkers are measurable indicators of a patient’s biological state or condition, CQAs are measurable indicators of a drug product’s quality. Traditionally, biomarker discovery efforts have been the domain of line functions related to translational medicine or correlative sciences whereas CQA discovery efforts have been the domain of line functions related to process development and manufacturing groups. For CAR-T cell therapy drug development, however, CQA and biomarker discovery efforts will need to integrate in order to yield the most comprehensive and holistic understanding of drug product safety and efficacy.
CAR-T cell manufacturing is considerably more challenging than that of traditional biologics such as monoclonal antibodies (MAbs). This is due to the much greater complexity of live cell products, incomplete understanding of their mechanisms of action, difficulties in product characterization and variability of the starting material. With respect to starting material, for example, most CAR-T cell therapy manufacturing protocols make use of autologous peripheral blood mononuclear cell (PBMC) concentrates collected by apheresis. In addition to lymphocytes, however, the apheresis product is often contaminated with variable quantities of red blood cells, monocytes, neutrophils, platelets, blast cells and myeloid derived suppressor cells. The frequency of these contaminating cells can have a significant impact on the overall success of the CAR-T cell manufacturing process [7]. As such, a rigorous analytical plan that can identify and monitor CQAs for the incoming apheresis starting material becomes central to the overall success of the CAR-T cell therapy manufacturing strategy.
Within the context of MAb manufacturing, cells are used as an expression system that secretes the target therapeutic product into their culture media. Alternatively, the therapeutic product is retained within the producer cell line and subsequently removed through mechanical and/or chemical means in a downstream purification process. Importantly, at some point in the biomanufacturing process of such biologics, the interdependence of a cellular expression system’s CQAs and those of the therapeutic product must and can be separated from one another [8,9]. That, however, is not possible with cell therapies such as CAR-Ts, because the cells themselves are the product that will be administered. Thus, end product CQAs and the bioprocess environment experienced by therapeutic cells are interdependent and require concomitant optimization, control and monitoring. This applies to the cell culture operations and the full manufacturing process from entry of raw materials through final product formulation. Industrial cell lines typically used to produce biologics, such as MAbs, exhibit substantially more physicochemical stability during bioprocessing than ‘biologically dynamic’ autologous therapeutic cells, such as the T cells used for CAR-T manufacturing, whose fate is influenced by their patient origins and the bioprocessing environment. One of the least understood end product CQAs of the CAR-T cell therapy drug product is potency.
The FDA defines potency as “the specific ability or capacity of the product, as indicated by appropriate laboratory tests or by adequately controlled clinical data obtained through the administration of the product in the manner intended, to effect a given result” [10]. Potency measurements are one component of the overall analytical characterization strategy necessary for product characterization testing, comparability studies and stability protocols, which are used to establish that a consistently manufactured product is administered during all phases of clinical investigation. Ideally, the potency assay will represent a drug product’s mechanism of action. Within the context of CD19-directed CAR-T therapies, the most commonly utilized potency assays are cytotoxicity, as measured via chromium release, and interferon gamma (IFN-γ) release as measured by ELISA. In both cases, the final patient-specific CAR-T cell therapy drug product is co-cultured with a cell line, such as K562 cells, that stably expresses the CAR-T-specific tumor antigen. Despite their extensive use as release assays for potency, neither the IFN-γ nor the cytotoxicity assay are predictive of overall clinical efficacy and safety [11,12]. This should come as no surprise as both assays fail to take into account the full diversity of T-cell phenotypes and functions. Ex vivo characterization of peripheral blood derived T cells using mass cytometry have shown that there are up to hundreds of thousands of unique T-cell phenotypes that possess a myriad of functions [13]. Although a comprehensive characterization of CAR-T cell therapy drug products has not been performed to date, it is presumed that the infused CAR-T cell product possesses extensively more functional attributes than IFN-γ and cytotoxicity.
Moreover, because the IFN-&gammaand cytotoxicity assays use contrived non-patient specific cell lines as surrogate tumor targets, the assays neglect to take into account the patient-specific tumor heterogeneity and the tumor microenvironment (TME) within which the infused CAR-T cell therapy drug product will operate. With respect to the TME, for example, several recent reports have shown that the potency of anti-tumor T cells is heavily modulated by co-infiltration of macrophages and other myeloid cell types and that these interactions play meaningful roles in patient survival [14,15]. These results highlight the importance of personalized immune response profiles when studying the factors CAR-T cell therapy drug product potency and overall clinical response.
Moving forward, it will be necessary to design potency assays that take into account the full spectrum of clinically relevant functions within the CAR-T cell therapy drug product and ideally, if possible, design these assays to be more reflective of the patient-specific tumor and TME. In fact, the FDA suggests that one assay will likely not be sufficient to measure the potency of cell therapy products, such as CAR-T cell therapies, that display complex mechanisms of action. In such a scenario, the FDA recommends developing multiple complementary assays that measure different product characteristics associated with quality, consistency and stability [10]. When used together and when results are correlated with a relevant biological activity, these complementary assays should provide an adequate measure of potency.
Manufacturing success, within the context of traditional drug development, is often a function of dose. If a manufacturing process is able to make a sufficient quantity of the final drug product for initial clinical testing and subsequent scale up and scale out at reasonable economies of scale, the manufacturing process is considered successful. For traditional biologics, the aim of manufacturing is clear: to design processes and cell lines that enable increased production titers of the drug product. For CAR-T cell therapies, however, the concept of dose and manufacturing success is less well defined. Manufacturing processes that yield more CAR-T cells are not necessarily better than processes that yield less overall numbers of CAR-T cells. Unlike standard therapeutic drugs, CAR-T cells have the potential to proliferate and persist long after adoptive transfer making the half-life of these T cells incalculable and their effects indefinite. One study estimated the half-life of retrovirally gene-modified T cells infused into patients to be at least >16 years [16]. By extension, results from many CAR T cell therapy trials have failed to identify a clear correlation between T-cell dose and clinical outcome [17]. For example, in a CD19 CAR-T cell therapy trial for CLL patients conducted at the University of Pennsylvania, wherein lentivirally transduced T cells expressing the CAR construct in association with a 41BB co-stimulatory domain were used, investigators reported an optimal anti-tumor response at a T cell dose 40 to 80 times lower than that infused into the other two patients reported in this cohort [18,19]. Similarly, in studies conducted at the Memorial Sloan Kettering Cancer Center (MSKCC), wherein retrovrially transduced T cells expressing the CAR construct in association with a CD28 co-stimulatory domain were used, the investigators noted better objective responses in a lower dose cohort when compared to patients treated at a three-fold higher CAR-T cell therapy drug product dose [20]. Therefore, based on currently published reports there does not appear to be a correlation between T cell dose and clinical outcome within a large-range of clinically meaningful treatment doses (2 × 105 to 3.1 × 107 CAR+ T cells/kg) [17]. Collectively, these finding suggest that clinical factors that go beyond absolute number of manufactured CAR-T cells, such as the patient’s tumor burden, are likely to play an important role in setting the limits of dose and by extension the concept of manufacturing success [17].
By extension, the traditional pharmacological notions of adsorption, distribution, metabolism and excretion (ADME) cannot be used to model the pharmacokinetics of CAR-T cells. Since these cells are usually infused intravenously, adsorption is clearly not an issue. Conversely, distribution, metabolism and excretion also apply, but need to be thoroughly reconsidered [21]. A recent study by Chapuis et al. nicely illustrates the complexity of dose and pharmacokinetics with respect to ‘living’ T cell immunotherapy drug products such as CAR-T therapies [22]. In this study, the authors applied high-throughput TCR Vß sequencing to identify individual T cell clonotypes within a polyclonal tumor-specific cytotoxic T cell (CTL) drug product. Using this approach, they were able to track individual clonotypes in vivo after infusion into patients and then deduce the pre-adoptive transfer (endogenous) frequencies of cells ultimately responsible for tumor regression. The authors found that in patients who achieved a durable complete response, the composition of the transferred CTLs was dominated by cells derived from a single T cell clonotype and that this clonotype was often present in the endogenous sample at less than 0.001%. These results suggest that the anti-tumor response is mediated by rare T cell populations that exhibit proliferative or survival advantages.
Integrated analytical strategy for identifying clinically relevant disease biomarkers & car-t drug product CQAs
As can be seen, the successful development of CAR-T cell therapies is currently hindered by our lack of comprehensive mechanistic knowledge about the drug product being tested, with respect to CQAs, and the patient-specific biomarkers that enable the CAR-T cell therapy drug product to be fully functional in vivo. Added to this complexity is the fact that CAR-T cell therapies blur many of the boundaries of traditional drug development. For example, the patient derived starting material (i.e., apheresis) would traditionally be considered a clinical sample and therefore the purview of translational medicine or correlative sciences groups within a pharmaceutical/biopharmaceutical organization. Yet, within the context of cell therapies, thorough understanding of the apheresis is fundamentally important for ensuring reproducible manufacturing and therefore CMC groups within a pharmaceutical/biopharmaceutical company have an equal claim to these samples for discovery purposes aimed at identifying CQAs related to manufacturing success. Likewise, the concept of manufacturing success is traditionally related to the ability of CMC groups to manufacture sufficient amounts of product to enable dosing of patients. Yet, as discussed, dose is often a function of the patient’s disease state (i.e., tumor burden, immune set point, etc.) and therefore determination of ‘manufacturing success’ is just as much a product of translational medicines/correlative sciences groups as it is for CMC.
In the face of this complexity, one possible solution is to implement a systems biology approach toward the identification of CQAs and biomarkers that are relevant for manufacturing and clinical success. This process requires coordinating clinical biomarker and manufacturing CQA efforts across the entire drug development process from preclinical discovery assays, to manufacturing and quality control release assays, to correlative studies and clinical readouts. Application of high-throughput assays that capture large amounts of quantitative information from limited amounts of clinical material becomes paramount, especially in early phase clinical trials.
Multiparametric platforms and assays that capture diverse sets of information include cell-based platforms to interrogate immune cell phenotype and function, nucleic acid, soluble or proteomic assay systems that allow for simultaneous and quantitative analyses of large numbers of analytes from a single small sample; high-throughput TCR sequencing strategies to understand the breadth of antitumor CAR-T cell responses and high-throughput serum based proteomic strategies to evaluate antitumor responses. This type of approach has already yielded several tangible success stories for CAR-T cell therapies. For example, one study that used a high-throughput and agnostic serum based proteomic platform on CAR-T cell treated patients that there may be epitope spreading following therapy indicative of a systemic tumor-specific adaptive immune response [23]. In another study, the authors were able to use various bioinformatics tools and prediction algorithms to identify new T cell epitopes, which enabled the identification of T cells that are reactive to these epitopes for further function testing of their target cell killing in vitro [24]. Finally, the use of agnostic cytokine analysis enabled the identification of IL-6 as a marker of severe cytokine release syndrome (CRS) in a subset of CAR-T cell therapy treated patients; this observation subsequently led to the use of tocilizumab treatment as an effective mitigation strategy for this potentially fatal syndrome [25].
The above cases argue for a model wherein broad, agnostic and quality-controlled analytical platforms are used in early phase clinical trials to comprehensively profile patient-specific clinical biomarkers and drug product-specific attributes that may be related to CQAs (Figure 2).
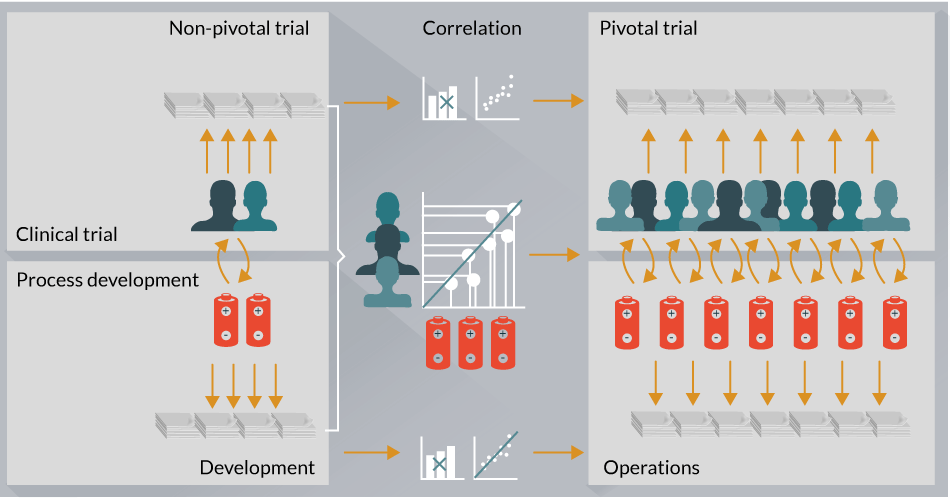
Figure 2: Proposed model of CAR-T cell therapy integrated analytical development strategy. CAR-T cell therapy drug development will require an analytical characterization strategy that enables for clinical biomarker and manufacturing CQA identification efforts in a systematic and integrated manner across the entire pharmaceutical and biopharmaceutical organization. Moreover, this process is likely to be iterative and enable cross talk between pivotal and non-pivotal trials.
Integration of these two data sets (i.e., patient-specific biomarkers and drug product-specific attributes) will enable generation of comprehensive hypotheses related to functional and mechanistic aspects of CAR-T therapies, which can then be tested prospectively in more advanced pivotal phase clinical trials (Figure 2). Such an approach has the potential to increase our understanding of patient and drug product-specific attributes that associate with overall safety and efficacy and will be an important driver for next generation manufacturing and clinical trial strategies aimed at scaling out CAR-T cell therapies.
Financial and competing interests disclosure
The author has no relevant financial involvement with an organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock options or ownership, expert testimony, grants or patents received or pending, or royalties. No writing assistance was utilized in the production of this manuscript.
References
1. Lim WA, June CH. The Principles of Engineering Immune Cells to Treat Cancer. Cell 168(4): 724–40.
CrossRef
2. Sadelain M. Chimeric antigen receptors: driving immunology towards synthetic biology. Curr. Opin. Immunol. 2016; 41: 68–76.
CrossRef
3. Sadelain M, Brentjens R, Riviere I. The promise and potential pitfalls of chimeric antigen receptors. Curr. Opin. Immunol. 2009; 21(2): 215–23.
CrossRef
4. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature 2017; 541(7637): 321–30.
CrossRef
5. Hey SP, Kesselheim AS. The FDA, Juno Therapeutics, and the ethical imperative of transparency. BMJ 2016; 354.
CrossRef
6. Highlights GN, Juno Halts Development of CAR T-Cell Cancer Immunotherapy Candidate JCAR015. Genetic Engineering and Biotechnology News, 2017.
Webiste
7. Stroncek DF, Ren J, Lee DW et al. Myeloid cells in peripheral blood mononuclear cell concentrates inhibit the expansion of chimeric antigen receptor T cells. Cytotherapy 2016; 18(7): 893–901.
CrossRef
8. Berkowitz SA, Engen JR, Mazzeo JR, Jones GB. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat. Rev. Drug Discov. 2012; 11(7): 527–40.
CrossRef
9. Carmen J, Burger SR, McCaman M, Rowley JA. Developing assays to address identity, potency, purity and safety: cell characterization in cell therapy process development. Regen. Med. 2011; 7(1): 85–100.
CrossRef
10. FDA, Draft Guidance for Industry: Potency Tests for Cellular and Gene Therapy Products. 2015.
Webiste
11. Lin-Gibson S, Rogers KC, Plant AL. Measurement Challenges for CAR-T Biomanufacturing: Highlights from a Meeting Sponsored by the National Institute of Standards and Technology (NIST). Hum. Gene Ther. Clin. Dev. 2016; 27(2): 66–8.
CrossRef
12. Mezher M. FDA Proposes New Databases to Monitor CAR T-Cell Safety Across INDs. Regulatory Affairs Professionals Society, 2016.
Website
13. Newell EW, Lin W. High-dimensional analysis of human CD8(+) T cell phenotype, function, and antigen specificity. Curr. Top. Microbiol. Immunol. 2014; 377: 61–84.
CrossRef
14. Hartmaier RJ, Charo J, Fabrizio D et al. Genomic analysis of 63,220 tumors reveals insights into tumor uniqueness and targeted cancer immunotherapy strategies. Genome Med. 2017; 9(1): 16.
CrossRef
15. Spranger, S, Luke JJ, Bao R et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell–inflamed tumor microenvironment in melanoma. Proc. Natl Acad. Sci. 2016; 113(48): E7759–68.
CrossRef
16. Scholler J, Brady TL, Binder-Scholl G et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 2012; 4(132): 132ra53.
CrossRef
17. Davila ML, Brentjens R. CAR Therapy for CLL: What are the Challenges? Hematol. Oncol. Clin. North Am. 2013; 27(2): 341–353.
CrossRef
18. Kalos M, Levine BL, Porter DL et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011; 3(95): 95ra73.
CrossRef
19. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011; 365(8): 725–33.
CrossRef
20. Brentjens RJ, Rivière I, Park JH et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011; 118(18): 4817–28.
CrossRef
21. Norelli M, Casucci M, Bonini C, Bondanza A et al. Clinical pharmacology of CAR-T cells: Linking cellular pharmacodynamics to pharmacokinetics and antitumor effects. Biochim. Biophys. Acta 2016; 1865(1): 90–100.
CrossRef
22. Chapuis AG, Desmarais C, Emerson R et al. Tracking the fate and origin of clinically relevant adoptively transferred CD8+ T cells in vivo. Sci. Immunol. 2017; 2(8).
CrossRef
23. Beatty GL, Haas AR, Maus MV et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2014; 2(2): 112–20.
CrossRef
24. Kreiter S, Vormehr M, van de Roemer N et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015; 520(7549): 692–6.
CrossRef
25. Grupp SA, Kalos M, Barrett D et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013; 368(16): 1509–18.
CrossRef
26. Jensen MC, Popplewell L, Cooper LJ et al. Anti-Transgene Rejection Responses Contribute to Attenuated Persistence of Adoptively Transferred CD20/CD19-Specific Chimeric Antigen Receptor Re-directed T Cells in Humans. Biol. Blood Marrow Transplant. 2010; 16(9): 1245–56.
CrossRef
27. Savoldo B, Ramos CA, Liu E et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Invest. 2011; 121(5): 1822–6.
CrossRef
28. Kochenderfer JN, Dudley ME, Feldman SA et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012; 119(12): 2709–20.
CrossRef
29. Kochenderfer JN, Wilson WH, Janik JE et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010; 116(20): 4099–102.
CrossRef
30. Brentjens RJ, Davila ML, Riviere I et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013; 5(177): 177ra38.
CrossRef
31. Davila ML, Riviere I, Wang X et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014; 6(224): 224ra25.
CrossRef
32. Kochenderfer JN, Dudley ME, Carpenter RO et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 2013; 122(25): 4129–39.
CrossRef
33. Lee DW, Kochenderfer JN, Stetler-Stevenson M et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015; 385(9967): 517–28.
CrossRef
34. Maude SL, Frey N, Shaw PA et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014; 371(16): 1507–17.
CrossRef
35. Pharma K. Kite Announces Positive Topline Primary Results of Axicabtagene Ciloleucel from First Pivotal CAR-T Trial in Patients with Aggressive Non-Hodgkin Lymphoma. February 28, 2017.
Website
36. Pharmaceuticals N. Novartis presents results from first global registration trial of CTL019 in pediatric and young adult patients with r/r B-ALL. 2016.
Website
37. Kroschinsky F, Stölzel F, von Bonin S et al. New drugs, new toxicities: severe side effects of modern targeted and immunotherapy of cancer and their management. Crit. Care 2017; 21(1): 89.
CrossRef
38. Suntharalingam G, Perry MR, Ward S et al. Cytokine Storm in a Phase 1 Trial of the Anti-CD28 Monoclonal Antibody TGN1412. N. Engl. J. Med. 2006; 355(10): 1018–28.
CrossRef
39. Lee DW, Gardner R, Porter DL et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014; 124(2): 188–95.
CrossRef
40. Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N. Engl. J. Med. 2011; 364(19): 1844–54.
CrossRef
41. Howard SC, Trifilio S, Gregory TK, Baxter N, McBride A. Tumor lysis syndrome in the era of novel and targeted agents in patients with hematologic malignancies: a systematic review. Ann. Hematol. 2016; 95(4): 563–73.
CrossRef
42. Maus MV, Levine BL. Chimeric Antigen Receptor T-Cell Therapy for the Community Oncologist. Oncologist 2016; 21(5): 608–17.
CrossRef
43. Zhu Y, Tan Y, Ou R et al. Anti-CD19 chimeric antigen receptor-modified T cells for B-cell malignancies: a systematic review of efficacy and safety in clinical trials. Eur. J. Haematol. 2016; 96(4): 389–96.
CrossRef
44. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016; 3: 16011.
CrossRef
Affiliations
Sadik H Kassim
Vice President, Process and
Analytical Development,
Mustang Bio
skassim@mustangbio.com

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.