Repurposing CRISPR-Cas systems as DNA-based smart antimicrobials
Cell Gene Therapy Insights 2017; 3(1), 63-72.
10.18609/cgti.2017.008
The molecular machines from CRISPR-Cas immune systems provide versatile platforms for programmable targeting of DNA, which is commonly used to edit eukaryotic genomes, transcriptomes and epigenomes. Recently, CRISPR-Cas systems have been repurposed as specific antimicrobials, given their ability to selectively target and degrade bacterial DNA. Endogenous Cas nucleases can be redirected against pathogenic bacterial chromosomes using self-targeting CRISPR arrays to drive highly specific programmed cell death through targeted DNA damage. Likewise, complete CRISPR-Cas systems can be delivered to bacteria responsible for infectious disease to drive the genesis of lethal DNA breaks. Here, we discuss the various CRISPR-Cas systems and their potential exploitation to create weaponized phages as vectors for effective and selective eradication of bacterial pathogens. This opens new avenues for infectious disease therapies and the engineering of microbiome composition, while addressing the challenges inherent to broad-spectrum antibiotics, enabling selective eradication of pathogens and preventing the selection for antibiotic resistance.
The repurposing of effector nucleases from clustered regularly interspaced short palindromic repeats (CRISPR) -Cas systems has fueled the genome editing revolution, with the ability of Cas9 to rapidly and specifically target DNA sequences of interest [1,2]. CRISPR-based technologies have been adapted for numerous uses in therapeutics, biotechnology, agriculture and basic research [3]. However, the use of these systems in their native context has been relatively underexploited in bacteria [4,5]. Despite their colloquial use as gene editing machines, CRISPR-Cas systems in nature function as adaptive immunity in bacteria and archaea [6]. These systems hinge on DNA-encoded [7], RNA-mediated [8], DNA-targeting processes [9,10] to fend off invasive genetic elements such as bacteriophages and plasmids. These systems were originally identified in E. coli in the 1980s [11], repeatedly observed in the genomes of many bacteria and most archaea in the early 2000s [12], and have been characterized in the past decade [13]. Recently, these systems have been repurposed to induce programmed cell death by redirecting effector nucleases towards the bacterial chromosome using self-targeting CRISPR spacers [14]. The relative paucity and inefficiency of DNA repair pathways render self-targeting CRISPR events lethal [15,16].
Whereas the discovery of CRISPR-Cas immune systems and their harnessing for genome editing is relatively recent [13], the historical use and abuse of broad spectrum antibiotics has led to the ongoing rise of antimicrobial resistance in pathogenic bacteria, creating a sizeable and rapidly rising concern for medicine. Furthermore, as our understanding of the nature and role of the human microbiome in health and disease advances, the concerns over the broad eradication of bacterial populations associated with humans are rising[17,18]. Altogether, these trends have created a need for the development of specific antimicrobials that selectively target pathogenic bacteria, enable survival of commensal and beneficial microbes and reduce the selective pressure on antibiotic resistance genes. Here, we review the use of CRISPR-Cas systems to selectively eradicate bacterial populations and discuss their potential use in medicine and other industries.
The unmet need for narrow spectrum antibacterials
Conventional antibiotics have been remarkably cost-effective and efficacious therapeutic agents for nearly eight decades. Historically, the majority of these broad-spectrum antibiotics are derived from five distinct functional classes of antibiotics: ß-lactams, cell wall/membrane inhibitors, metabolism inhibitors, protein synthesis inhibitors and RNA/DNA synthesis inhibitors [19].This is the product of reduced investment by large pharmaceutical companies, resulting in a lack of novel antibiotics, particularly for gram-negative pathogens [20]. Conventional antibiotics may now be facing rapid obsolescence due to the well-publicized emergence and rapidly increasing dissemination of antibiotic resistance. Arguably, their broad prescription in human medicine, and also nearly ubiquitous use and abuse in agriculture (for both crops and animals) has created an urgent need to develop alternatives, to both provide enhanced antimicrobials and also counter the rise of antibiotic resistance.
In parallel, the relatively recent elucidation of the human microbiome [21] and its role in human health affords a new and compelling rationale to move away from the ‘scorch the earth’ approach underlying broad spectrum antibiotics. The past decade has laid the foundation for a human microbiome renaissance, unearthing the many beneficial bacteria that inhabit various human tissues, organs and epithelia, and studying their impact on human health. Bacterial dysbiosis has been implicated in a number of major human diseases such as diabetes, obesity, cancer, gastrointestinal disorders, neurological disorders [22] and infectious diseases [23]. One noteworthy example is the ability of the healthy human gut microbiota to prevent the onset of Clostridium difficile infection (CDI), and colonization resistance afforded by fecal microbiota transplantations (FMTs) [24]. Several studies have shown that the use of broad-spectrum antibiotics in healthcare settings disrupts the normal gut flora and creates an environment amenable to CDI onset, persistence and recurrence [25,26]. As our understanding of microbiomes advances, and dedicated studies identify the beneficial and pathogenic bacteria involved in various human diseases, there is a need for technologies that selectively allow the precise removal of infectious disease agents. While additions and replacements using ‘healthy donors’ are a potential solution, preventing recurrence will rely on selective removal of undesirable bacteria to preserve the rest of the microbiome. Furthermore, therapeutic regulatory processes, safety, manufacturing and consistent and predictable efficacy favor selective therapies rather than brute force colonization and wholesale formulations. Thus, next-generation antimicrobials that selectively target specific bacteria offer desirable opportunities.
CRISPR-based antibacterial technologies
By nature, CRISPR-Cas systems provide sequence-specific and efficacious targeting and destruction of invasive nucleic acids. Targeting of chromosomal sequences by CRISPR-Cas immune systems in bacteria equates to an autoimmune disease that is highly cytotoxic [16]. Indeed, seminal observations exploring CRISPR-Cas systems with self-targeting spacers have shown that bacterial chromosome targeting by Cas effector nucleases is lethal [15,27,28]. Exploiting this concept, it was established that re-directing native Type I CRISPR-Cas immune systems in bacteria provided potent and programmable killing of bacteria in vitro[29]. Shortly thereafter, studies showed that CRISPR-Cas systems could be engineered to drive the removal of pathogenic bacteria in vivo [30,31]. These studies provided a basis for the elimination of select genotypes from bacterial populations, and the selection of harmful DNA removal by screening for antibiotic resistance gene removal, and the loss of expendable genomic islands [32]. This body of work illustrates the potential of CRISPR-based technologies for the selective removal of pathogens, and the genesis of desirable genotypes that could compete with pathogenic bacteria.
In nature, CRISPR-Cas systems are a diverse collection of nucleic acid targeting molecular machines [33,34]that hinge either on a multi-protein effector complex (class 1), such as Cascade-Cas3 or single effector nuclease (class 2), such as Cas9. Each class contains multiple types, with types I and III within class 1 the most diverse and dominant in nature, followed by type II within class 1 (Figure 1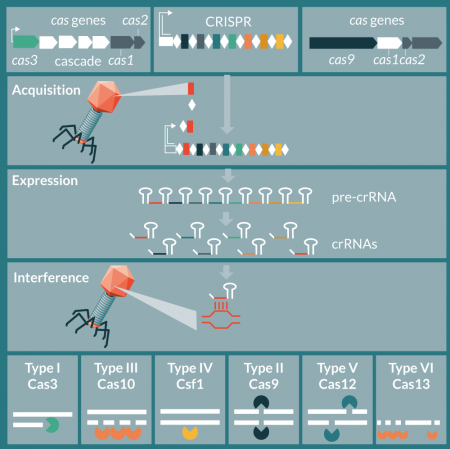
Engineering & delivery of CRISPR-based antibacterial payloads
Conceptually, there are two means to repurpose CRISPR-Cas systems as sequence-specific antimicrobials: co-opting native systems in pathogenic bacteria to trigger a lethal autoimmune response or deliver a complete system de novo to target the bacterial chromosome. For the former, the sole need is to design and deliver either a guide RNA (containing a processed spacer sequence defining the target) or a synthetic CRISPR array mimicking the native locus, carrying a self-targeting spacer [29]. For the latter, there is a need to deliver both the CRISPR array and the Cas machinery to generate the crRNAs that will guide exogenous Cas nucleases towards the engineered target (Figure 2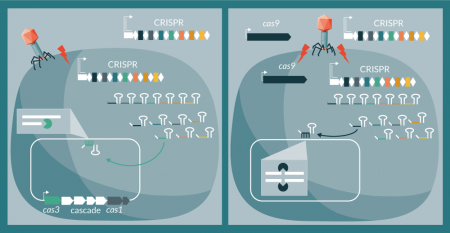
Conveniently, the widespread occurrence of type I systems afford opportunities to just deliver CRISPR arrays to pathogenic bacteria that carry these loci. Conversely, streamlined type II systems are more portable alternatives that can be more conveniently engineered. For either system, it is necessary to know the basic molecular processes that drive targeting in order to design guide RNAs accordingly. These parameters include the sequence, composition and boundaries of the processed crRNA, selecting a spacer sequence targeting a proto-spacer associated with a PAM, and choosing a particular locus of interest in the target organism, which has the ideal specificity as to ensure selective targeting of a particular bacterial genotype [53,54]. Consequently, it is paramount to have extensive genomic information about the targeted species to ensure that all, but only strains within this species or genotype are targeted. Of course, the very nature of CRISPR arrays enables multiplexed targeting by the concurrent provision of multiple spacers in a single CRISPR array. In terms of efficiency, the ability to concurrently drive cytotoxicity by using multiple targeting spacers enables an increase in killing levels, due to the multiplicity of escape hurdles.
In terms of packaging and delivery, bacteriophages provide convenient, portable, specific and high-capacity vectors that drive host-specific targeting and efficient delivery of DNA into a bacterial host. Whereas classical phage therapy relies on the ability of the phage to take over the host to achieve viral replication and host death, using CRISPR-based targeting relies on the host biology. Indeed, in the case of repurposing endogenous CRISPR-Cas systems, CRISPR array transcription is the sole requirement, since natively and constitutively expressed Cas effector nucleases can be harnessed. Furthermore, this strategy would enable the use of either lytic or lysogenic phages, broadening the delivery options. While the utility of CRISPR-based killing with lysogenic phages is obvious, CRISPR-weaponized obligate lytic phages may benefit from improved cytotoxicity, rather than relying on the viral replication life cycle alone.
To date, two studies have illustrated the in vivo efficacy potential of CRISPR-weaponized phage, one showing temperate phage NM1 targeting of Staphylococcus aureus [31],and the other showing filamentous phage M13 targeting of Escherichia coli [30]. Another recent study has shown how lambda phage can deliver CRISPR arrays and exploit endogenous Type I CRISPR-Cas systems to (re-)sensitize Escherichia coli strains to antibiotics, with potential applications in treating medical surfaces and/or personnel [55].
By nature, bacteriophage are designed to inject viral DNA into the host cell specifically, and efficiently, so using the as delivery vehicles is logical. Ironically, this also constitutes virus-driven repurposing of an antiviral mechanism to kill the host, rather than enable viral resistance using a bacterial CRISPR-Cas immune system. Importantly, the phage-based delivery of CRISPR arrays affords three levels of specificity, with: (1) selective adhesion and injection of viral DNA into the bona fide host; (2) CRISPR-array and guide RNA compatibility only with the corresponding Cas machinery; (3) designer-dependent selective targeting of sequences complementary to the engineered CRISPR spacer. Of course, there are no off-target concerns to other bacterial genera, species and strains, nor obviously to human cells. Notwithstanding the ease with which genome editing is now perceived in general, the synthetic biology expertise necessary to engineer bacteriophage is still non-trivial, and requires understanding of phage genome malleability, engineering synthetic CRISPR arrays and generating a host-phage system that will yield functional virions.
Translational Insight
As with any rising technology, the therapeutic implementation of a novel class of antimicrobials requires a series of resources-intensive studies to ensure efficacy and safety, as well as providing the necessary foundation for manufacturing processes and regulatory approval. Classical IND-enabling studies encompassing pharmacokinetic/pharmacodynamics (PK/PD), absorption, distribution, metabolism and excretion (ADME) and non-GLP toxicology will be required. While the studies to date provide intriguing and compelling proof of concept for CRISPR-based specific antimicrobials, there is a need to provide the necessary in vivo data to advance toward the clinic in treating human infectious disease.
A primary driver of clinical implementation is ensuring the safety of novel therapeutics, necessitating the classical battery of animal in vivo studies for toxicity, biological availability and distribution, and ultimately dosage and efficacy. This is a key advantage of using CRISPR-Cas systems in their native bacterium, as the potential for off-target activity in human cells is virtually non-existent. With regards to delivery of CRISPR-Cas components to bacteria, the route of administration will vary depending on the site of infection, with intestinal (oral), skin (topical), lung (aerosol), blood (intravenous), epithelial (UTI) and tissue (local injection) delivery options in play.
In this regard, bacteriophage-based vectors are particularly promising for pairing with CRISPR technology as phage therapies have an established, albeit short, track record of safety in human clinical trials [56]. As with any phage-driven therapy, engineering, formulation, selection and blending of cocktails, and manufacturing of bacterial viruses each require dedicated work streams to eventually formulate a stable therapeutic. In particular, it will be important to select bacteriophages with sufficiently broad host range against a target species. Being able to find phages with naturally broad host range would be helpful, and some efforts are also underway to rationally expand structural features of phage particles to render them able to more promiscuously target hosts. And while the process of phage manufacturing is relatively straightforward, there are often manufacturing restrictions in the western world for phage products, requiring dedicated facilities to handle replicative phage and enhanced phage products. Beyond phage, there are opportunities to also exploit learnings from other CRISPR-based technologies, notably genome editing, and benefit from alternative delivery formats such as ribonucleoproteins, cell-penetrating peptides and future delivery vehicles. As the technology advances across the application field, CRISPR-based antimicrobials will also benefit from progress related to specificity, Cas engineering and optimization, guide selection and design and other technical improvements. While on- versus off-targeting may need to be improved, an advantage of prokaryotic applications is fewer concerns about specificity given the much smaller genome sizes (reducing the probabilistic concerns about off-targeting by several orders of magnitude), and the intended outcome (cell death).
Altogether, most of these challenges have individually been addressed before, but perhaps not in combination, illustrating the challenges inherent to CRISPR-based technologies to be delivered by bacteriophage for infectious disease. It will be interesting to monitor the balance between timeliness and comprehensiveness as this technology advances to the clinic, and ascertain the level of urgency with which investors and regulatory agencies will fuel and enable progress, respectively. Furthermore, it will be intriguing to gauge whether there is a technological push to exploit CRISPR-based antimicrobials to address orphan infectious diseases (e.g., carbapenem-resistance enterobacteriaceae), and the rise of antibiotic resistance, or a market pull to generate specific antimicrobials that enable specific removal of undesirable genotypes within microbiomes, as the bacteria associated with human diseases are identified. Of course, CRISPR-based antimicrobials also hold tremendous potential for implementation in food, biotechnology and agriculture (both crops and animals), and future needs will determine whether human therapeutics remain the primary driver of commercialization.
Acknowledgements
The authors gratefully acknowledge their colleagues and collaborators for their valued insights into CRISPR-Cas systems in general and antimicrobial applications in particular, as well as financial support from Locus Biosciences.
Financial and competing interests disclosure
The authors are inventors on several patents related to various uses of CRISPR-based technologies. Rodolphe Barrangou is a co-founder and SAB member of Intellia Therapeutics and Locus Biosciences. David G Ousterout is an officer and investigator of Locus Biosciences. No writing assistance was utilized in the production of this manuscript.
References
1. Pennisi E. The CRISPR craze. Science 2013; 341: 833–6.
CrossRef
2. Ledford H. CRISPR, the disruptor. Nature 2015; 522: 20–4.
CrossRef
3. Barrangou R, Doudna JA. Applications of CRISPR technologies in research and beyond. Nat. Biotechnol. 2016; 34: 933–41.
CrossRef
4. Barrangou R, van Pijkeren JP. Exploiting CRISPR-Cas immune systems for genome editing in bacteria. Curr. Opin. Biotechnol. 2016; 37: 61–8.
CrossRef
5. Selle K, Barrangou R. Harnessing CRISPR-Cas systems for bacterial genome editing. Trends Microbiol. 2015; 23: 225–32.
CrossRef
6. Marraffini LA. CRISPR-Cas immunity in prokaryotes. Nature 2015; 526: 55–61.
CrossRef
7. Barrangou R, Fremaux C, Deveau H et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007; 315: 1709–12.
CrossRef
8. Brouns SJ, Jore MM, Lundgren M et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 2008; 321: 960–4.
CrossRef
9. Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 2008; 322: 1843–5.
CrossRef
10. Garneau JE, Dupuis ME, Villion M et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010; 468: 67–71.
CrossRef
11. Ishino Y, Shinagawa H, Makino K et al. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987; 169: 5429–33.
CrossRef
12. Mojica FJ, Diez-Villasenor C, Soria E et al. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol. Microbiol. 2000; 36: 244–6.
CrossRef
13. Sontheimer EJ, Barrangou R. The Bacterial Origins of the CRISPR Genome-Editing Revolution. Hum. Gene Ther. 2015; 26: 413–24.
CrossRef
14. Beisel CL, Gomaa AA, Barrangou R. A CRISPR design for next-generation antimicrobials. Genome Biol. 2014; 15: 516.
CrossRef
15. Bikard D, Hatoum-Aslan A, Mucida D et al. CRISPR interference can prevent natural transformation and virulence acquisition during in vivo bacterial infection. Cell Host Microbe 2012; 12: 177–86.
CrossRef
16. Vercoe RB, Chang JT, Dy RL et al. Cytotoxic chromosomal targeting by CRISPR/Cas systems can reshape bacterial genomes and expel or remodel pathogenicity islands. PLoS Genet. 2013; 9: e1003454.
CrossRef
17. Langdon A, Crook N, Dantas G. The effects of antibiotics on the microbiome throughout development and alternative approaches for therapeutic modulation. Genome Med. 2016; 8: 39.
CrossRef
18. Wischmeyer PE. Are we creating survivors…or victims in critical care? Delivering targeted nutrition to improve outcomes. Curr. Opin. Crit. Care 2016; 22: 279–84.
CrossRef
19. Bush K, Page MG. What we may expect from novel antibacterial agents in the pipeline with respect to resistance and pharmacodynamic principles. J. Pharmacokinet. Pharmacodyn. 2017; doi:10.1007/s10928-017-9506-4 (Epub ahead of print).
CrossRef
20. Norrby SR, Nord CE, Finch R et al. Lack of development of new antimicrobial drugs: a potential serious threat to public health. Lancet Infect. Dis. 2005; 5: 115–9.
CrossRef
21. Human Microbiome Project C. Structure, function and diversity of the healthy human microbiome. Nature 2012; 486: 207–14.
CrossRef
22. Fung TC, Olson CA, Hsiao EY. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 2017; 20: 145–55.
CrossRef
23. Petersen C, Round JL. Defining dysbiosis and its influence on host immunity and disease. Cell Microbiol. 2014; 16: 1024–33.
CrossRef
24. Bakken JS, Borody T, Brandt LJ et al. Treating Clostridium difficile infection with fecal microbiota transplantation. Clin. Gastroenterol. Hepatol. 2011; 9: 1044–9.
CrossRef
25. Lessa FC, Winston LG, McDonald LC et al. Burden of Clostridium difficile infection in the United States. N. Engl. J. Med. 2015; 372: 2369–70.
CrossRef
26. Johnson S, Louie TJ, Gerding DN et al. Vancomycin, metronidazole, or tolevamer for Clostridium difficile infection: results from two multinational, randomized, controlled trials. Clin. Infect. Dis. 2014; 59: 345–54.
CrossRef
27. Jiang W, Bikard D, Cox D et al. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013; 31: 233–9.
CrossRef
28. Paez-Espino D, Morovic W, Sun CL et al. Strong bias in the bacterial CRISPR elements that confer immunity to phage. Nat. Commun. 2013; 4: 1430.
CrossRef
29. Gomaa AA, Klumpe HE, Luo ML et al. Programmable removal of bacterial strains by use of genome-targeting CRISPR-Cas systems. MBio 2014; 5: e00928-13.
CrossRef
30. Citorik RJ, Mimee M, Lu TK. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol. 2014; 32: 1141–5.
CrossRef
31. Bikard D, Euler CW, Jiang W et al. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol. 2014; 32: 1146–50.
CrossRef
32. Selle K, Klaenhammer TR, Barrangou R. CRISPR-based screening of genomic island excision events in bacteria. Proc. Natl Acad. Sci. USA 2015; 112: 8076–81.
CrossRef
33. Makarova KS, Wolf YI, Alkhnbashi OS et al. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015; 13: 722–36.
CrossRef
34. Barrangou R. Diversity of CRISPR-Cas immune systems and molecular machines. Genome Biol. 2015; 16: 247.
CrossRef
35. Makarova KS, Koonin EV. Annotation and Classification of CRISPR-Cas Systems. Methods Mol. Biol. 2015; 1311: 47–75.
CrossRef
36. Makarova KS, Wolf YI, Alkhnbashi OS et al. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015; 13: 722–3.
CrossRef
37. Makarova KS, Zhang F, Koonin EV. SnapShot: Class 2 CRISPR-Cas Systems. Cell 2017; 168: 328–328 e1.
CrossRef
38. Makarova KS, Zhang F, Koonin EV. SnapShot: Class 1 CRISPR-Cas Systems. Cell 2017; 168: 946–946 e1.
CrossRef
39. Deveau H, Barrangou R, Garneau JE et al. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 2008; 190: 1390–400.
CrossRef
40. Horvath P, Romero DA, Coute-Monvoisin AC et al. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J. Bacteriol. 2008; 190: 1401–12.
CrossRef
41. Mojica FJ, Diez-Villasenor C, Garcia-Martinez J et al. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 2009; 155: 733–40.
CrossRef
42. Sternberg SH, Redding S, Jinek M et al. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014; 507: 62–7.
CrossRef
43. Sinkunas T, Gasiunas G, Fremaux C et al. Cas3 is a single-stranded DNA nuclease and ATP-dependent helicase in the CRISPR/Cas immune system. EMBO J. 2011; 30: 1335–42.
CrossRef
44. Sinkunas T, Gasiunas G, Waghmare SP et al. In vitro reconstitution of Cascade-mediated CRISPR immunity in Streptococcus thermophilus. EMBO J. 2013; 32: 385–94.
CrossRef
45. Hochstrasser ML, Taylor DW, Bhat P et al. CasA mediates Cas3-catalyzed target degradation during CRISPR RNA-guided interference. Proc. Natl Acad. Sci. USA 2014; 111: 6618–23.
CrossRef
46. Huo Y, Nam KH, Ding F et al. Structures of CRISPR Cas3 offer mechanistic insights into Cascade-activated DNA unwinding and degradation. Nat. Struct. Mol. Biol. 2014; 21: 771–7.
CrossRef
47. Mulepati S, Bailey S. In vitro reconstitution of an Escherichia coli RNA-guided immune system reveals unidirectional, ATP-dependent degradation of DNA target. J. Biol. Chem. 2013; 288: 22184–92.
CrossRef
48. Barrangou R, Dudley EG. CRISPR-Based Typing and Next-Generation Tracking Technologies. Annu. Rev. Food Sci. Technol. 2016; 7: 395–411.
CrossRef
49. Yin S, Jensen MA, Bai J et al. The evolutionary divergence of Shiga toxin-producing Escherichia coli is reflected in clustered regularly interspaced short palindromic repeat (CRISPR) spacer composition. Appl. Environ. Microbiol. 2013; 79: 5710–20.
CrossRef
50. Shariat N, Timme RE, Pettengill JB et al. Characterization and evolution of Salmonella CRISPR-Cas systems. Microbiology 2015; 161: 374–86.
CrossRef
51. Andersen JM, Shoup M, Robinson C et al. CRISPR Diversity and Microevolution in Clostridium difficile. Genome Biol. Evol. 2016; 8: 2841–55.
CrossRef
52. Rollins MF, Schuman JT, Paulus K et al. Mechanism of foreign DNA recognition by a CRISPR RNA-guided surveillance complex from Pseudomonas aeruginosa. Nucleic Acids Res. 2015; 43: 2216–22.
CrossRef
53. Briner AE, Barrangou R. Guide RNAs: A Glimpse at the Sequences that Drive CRISPR-Cas Systems. Cold Spring Harb. Protoc. 2016; 2016: pdb top090902.
CrossRef
54. Briner AE, Barrangou R. Deciphering and shaping bacterial diversity through CRISPR. Curr. Opin. Microbiol. 2016; 31: 101–8.
CrossRef
55. Yosef I, Manor M, Kiro R et al. Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc. Natl Acad. Sci. USA 2015; 112: 7267–72.
CrossRef
56. Vandenheuvel D, Lavigne R, Brussow H. Bacteriophage Therapy: Advances in Formulation Strategies and Human Clinical Trials. Annu. Rev. Virol. 2015; 2: 599–618.
CrossRef
57. Hale CR, Zhao P, Olson S et al. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 2009; 139: 945–56.
CrossRef
58. Gasiunas G, Barrangou R, Horvath P et al. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl Acad. Sci. USA 2012; 109: E2579–86.
CrossRef
59. Jinek M, Chylinski K, Fonfara I et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012; 337: 816–21.
CrossRef
60. Zetsche B, Gootenberg JS, Abudayyeh OO et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015 163: 759–71.
CrossRef
61. Shmakov S, Abudayyeh OO, Makarova KS et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol. Cell 2015; 60: 385–97.
CrossRef
Affiliations
Rodolphe Barrangou1,2# and David G. Ousterout 2
1 Department of Food, Bioprocessing and Nutrition Sciences; North Carolina State University; Raleigh, NC 27695, USA; . rbarran@ncsu.edu; phone: (919) 513-1644.
2 Locus Biosciences, 7020 Kit Creek Road, Suite 210, Research Triangle Park, NC 27709, USA
# Corresponding author: rbarran@ncsu.edu

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.