Key steps to industrialize your process and de-risk your pathway to commercialization
Cell & Gene Therapy Insights 2020; 6(3), 469–479
10.18609/cgti.2020.056
In order to successfully manufacture clinical-grade GMP products, it’s important to take a step by step approach towards their development.
The first step, what we term the manufacturability assessment or diagnosis to de-risk manufacturing, focuses on establishing the baseline process, and identifying the major risks involved in our clients’ manufacturing process considering GMP guidelines. This step will help to define the focus and scope of the next step (i.e. development step).
Once there is a good understanding of the scope, then we move to development step and designing experiments centered around process optimization based on the manufacturing design considerations and critical quality attributes. We focus on different challenges, for example scale up or scale out, efficiency and yield, optimization of unit operations (for example, initial starting cell isolation), sensitivity and robustness of the process, development of appropriate analytical methods and assays, implementing appropriate in-process control characterizations in the process, and cell harvest and banking strategy. In addition, it’s important to evaluate the raw materials involved in the processes to make sure they fit the GMP guidelines and compliance.
We include our Manufacturing Science and Technology Team (MSAT) early on, even during development, to make sure this understanding of manufacturing design considerations is properly implemented into the process development strategy. Once the manufacturing process is robust and reproducible, we work with our MSAT to transfer this process into a GMP manufacturing setting.
Some of the best practices we recommend in the development of GMP manufacturing processes, to meet clinical and commercial needs, are highlighted in this Figure 1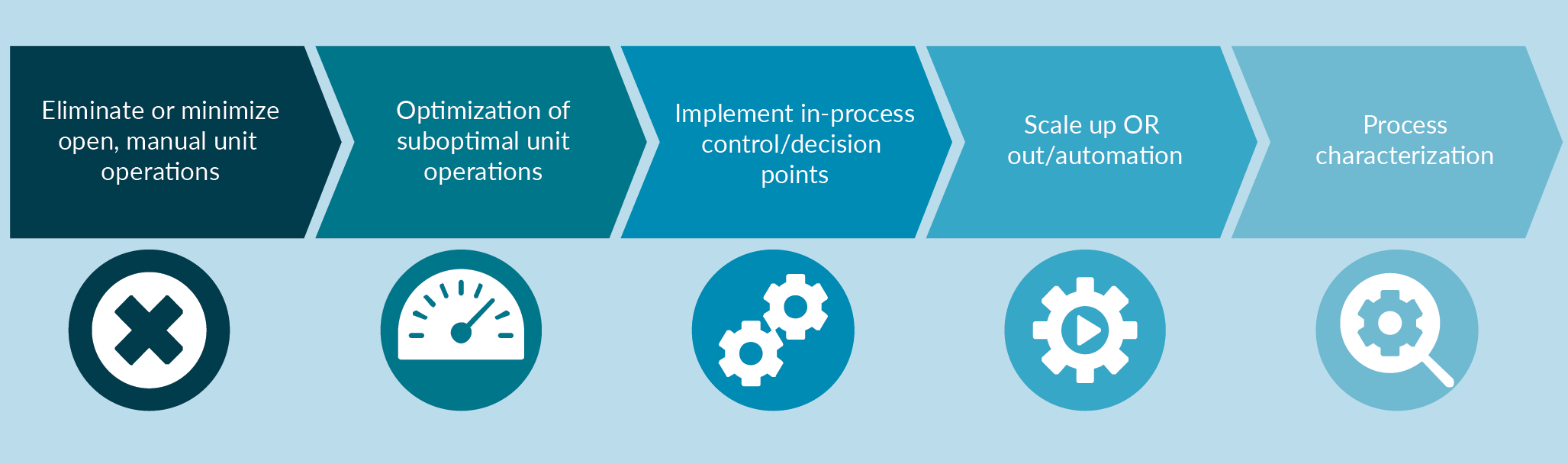
Case study 1: manufacturing of clinical-grade cell therapy products from iPSCs
This first case study pertains to a directed differentiation process starting from human induced pluripotent stem cells (iPSCs), which is a highly complex process that requires very accurate control of each step. There are several factors involved in the manufacturing process including the quality of starting material, cellular microenvironment, cell–cell interaction, signaling factors, and other important parameters.
The entire process can be divided into four major steps:
- Manufacturing of GMP-grade iPSCs;
- Expansion of iPSCs to create sufficient number of cells prior to the differentiation;
- Directed differentiation process;
- Characterization and testing of the final product (and intermediate materials) depending on the direct differentiation process.
For this particular process, some of the best practices we have implemented during our process development and optimization steps include the development of a robust cell culture system for generation of human iPSCs and expansion of pluripotent stem cells including iPSCs and human embryonic stem cells (hESCs) as well as development of bioreactor protocols for expansion pluriptent stem cells in 3D.
Our focus has been on establishing a high quality iPSC using our GMP manufacturing process and to that end, developing a comprehensive iPSC characterization platform. Having established both 2D and 3D bioreactor systems, this enabled directed differentiation of iPSCs into different lineages. Depending on the application, different strategies can be taken to optimize the manufacturing process, some of which are highlighted below.
| TABLE 1 Lonza generated multiple fully characterized iPSC lines. | ||||
|---|---|---|---|---|
| Assay | Objective | Evaluation criteria | Category | Tested iPSC line |
| Pluripotency markers | Identity and purity | SSEA-4 >70%, Tra-1-60 >70%, Tra-1-81 >70%, Oct3/4 >70%, Purity: CD34 <5% | Release assay | All lines |
| Karyotype analysis | Safety | 46, XX or 46, XY | Release assay | All lines |
| Mycoplasma testing | Safety | Negative | Release assay | All lines |
| Sterility testing | Safety | Negative | Release assay | All lines |
| Endotoxin testing | Safety | Standard QC release (<0.5 EU/ml) | Release assay | All lines |
| Vector clearance | Safety | No trace of episomal plasmid DNA detected | Release assay | All lines |
| STR genotyping | Purity and identity | STR Profile of starting population and iPSC line are identical | Release assay | Al lines |
| Cell count and viability | Viability | % viability >50; minimum cell number/vial | Release Assay | All lines |
| Viral panel testing | Safety | Standard MCB Release Panel | Release Assay | LiPSC-GR1.1 |
| Characterization assays | ||||
| EB formation | Identity and potency | Detection of at least one marker per germ layer | FIO* | All lines |
| Gene array analysis | Identity | Clustering with established hPSCs | FIO* | All lines |
| Colony morphology | Identity and purity | Characteristic morphology of culture/colonies; lack of sponta- neously differentiated cells | FIO* | All lines |
| Post-thaw plating | Thawing efficiency and viability | 20+ colonies/vial (after 7 days or 50% confluency) | FIO* | All lines |
| HLA typing | Identity | HLA-A, B, C, DRB1 and DQB1Type | FIO* | All lines |
| CGH+SNP microarray | Identity | Amplifications and/ or deletions of specific genes | FIO* | LiPSC-GR1.1 and ER2.2 |
| Whole genome sequencing | Identity | HiSeq X Human Whole Genome Sequence | FIO* | LiPSC-GR1.1 and ER2.2 |
| (Baghbaderani et al. 2016; Stem Cell Reviews and Reports ). | ||||
L7TM cell culture system: a defined & cGMP compliant cell culture system for manufacturing of human PSCs
In order to establish a robust and reproducible cell therapy manufacturing process, it is essential to use a high quality starting material, in this case iPSCs, and with that in mind we established a GMP manufacturing process using a defined and cGMP-compliant L7TM cell culture system. This has enabled the generation of iPSCs under GMP guidelines, the details of which have been published previously [1].
Our group has extensively focused on the development of a series of analytical methods that are required for release and characterization of iPSCs, as shown in Table 1 [2].
In the next step we have shown that iPSCs generated in our cell culture system and manufactured under GMP can be used in the directed differentiation process for different clinical applications and hold the potential to differentiate into all three main lineages: ectoderm, mesoderm and endoderm.
To help address some of the current manufacturing challenges associated with the use of traditional, open and manual 2D unit operations (which may lead to major quality, quantity and efficiency concerns), we considered it essential to establish a computer-controlled 3D bioreactor system. In order to develop a 3D bioreactor based process, we focused on several important process parameters, including inoculation condition, hydrodynamics of the bioreactor, process control, expansion, feeding strategies.
We took a step by step approach towards the scale up of the iPSC-derived processes, from 2D cell culture systems into 3D suspension; first moving into a spinner flask, and then into a larger scale computer controlled bioreactor.
We demonstrated our approach towards scale up and establishing a 3D process by focusing on iPSC differentiation into cardiomyocytes using a protocol reported in the literature [3]. First, we optimized the differentiation process in 2D conditions before moving to 3D. Next, we optimized the cell culture and directed differentiation conditions in spinner flask to enable the transition of the process into a 3D environment. We demonstrated that the iPSCs derived under GMP conditions can be differentiated into cardiomyocytes in 3D as supported by cellular characterization studies. We also evaluated various process parameters, including aggregate size distribution in 3D, as well as cell metabolite and nutrient composition and used these parameters to optimize the process and control critical quality attributes of the process. Some of the process variables and target criteria including the cell viability, key surface marker expression are exhibited in Figure 2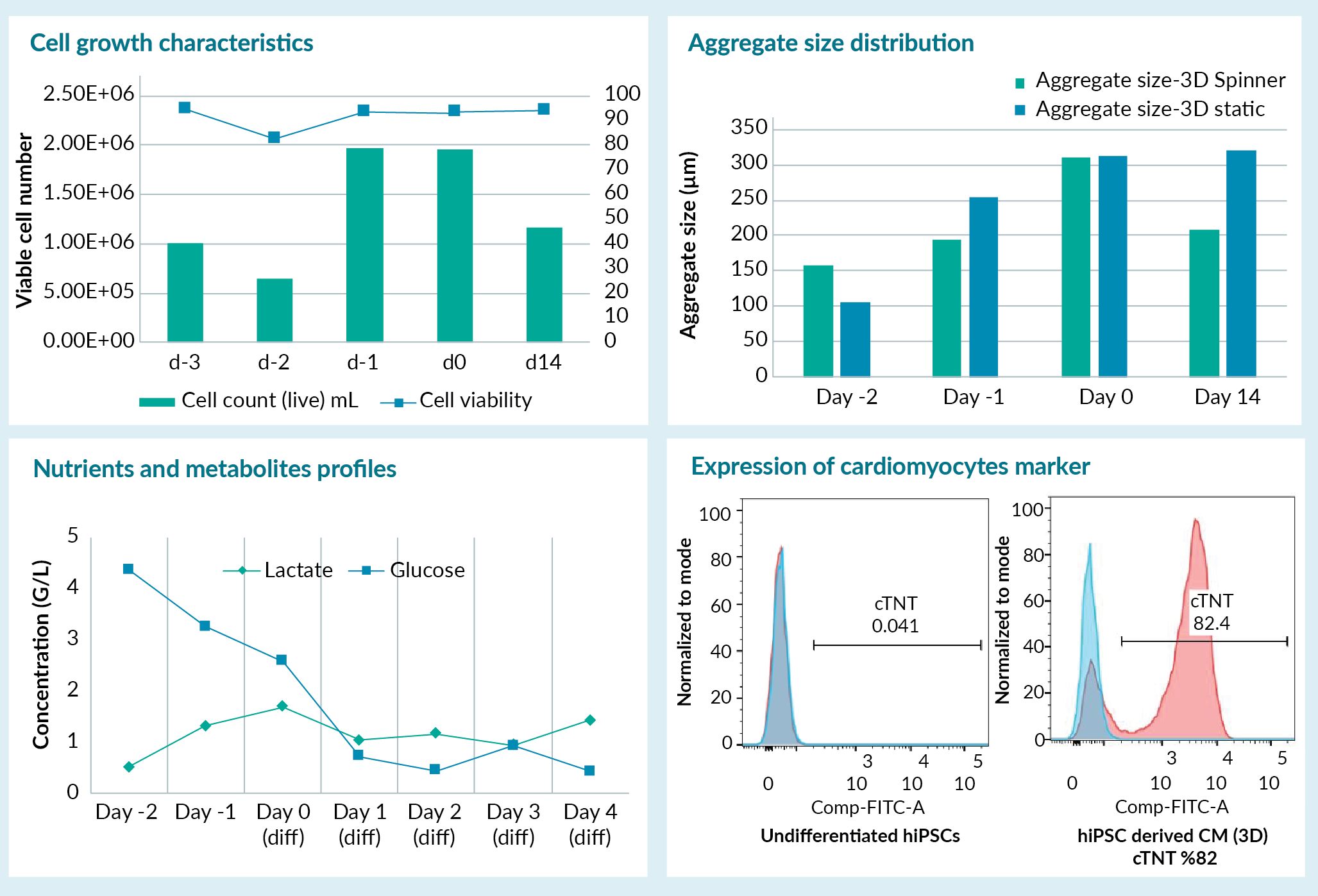
From a scale-up perspective, we then focused on incorporating novel bioreactor technologies into the manufacturing process and direct differentiation moving from small uncontrolled 3D, spinner flasks into larger scale computer controlled bioreactors. For instance, we used computational fluid dynamics (CFD) modelling in scale up [4]. Using this method we took into consideration the geometry of the vessel, and more accurately, evaluated the hydrodynamics of the bioreactor, allowing scale up of the process from small spinner flasks into the larger scale 3D bioreactor.
In conclusion, we have been able to show that iPSCs generated under cGMP conditions could be differentiated into all three lineages successfully in 2D and 3D, and these processes can be established and scaled up as needed depending on the mode of culture [3]. Our most recent publication details the CFD modelling we’ve used for the development of scalable cell therapy manufacturing process [4].
Case study 2: manufacturing of clinical-grade CAR-T product
For a CAR-T-based application, the manufacturing process is complex involving many unit operations in different combinations. A current challenge with this type of autologous cell manufacturing process is that many of these unit operations are open and manual, and use uncontrolled culture conditions, increasing risks of contamination and driving up costs of manufacturing. To fundamentally address these challenges, automation may be a feasible approach to replace open manual steps with more automated approaches. The automation can be a modular (i.e., focusing on one or more unit operations of the manufacturing process, for instance cell isolation step) or end-to-end approach (i.e., focusing on all of the unit operations in the entire manufacturing process from isolation to fill and finish), the latter of which would be our preference. Some of the unit operations that may need specific attention include apheresis product processing and cell isolation, ficollation or density based selection, magnetic selection of specific cell types, and/or buffer exchange into culture medium. For instance, Cell isolation is the first step in the manufacturing process, and is often a manual unit operation. Our recommended approach is to move this manual unit operation into automated closed system modalities to remove the inherent risks associated with the operator dependent manipulations and reduce labor-intensive procedures. However, the current most prevalent approach to automation is modular, owing to a lag in development of appropriate end-to-end, fully closed, automated platform technologies.
In addition to automation, process optimization and elimination or reduction of suboptimal unit operations with more robust and optimized process steps require specific attention. Figure 3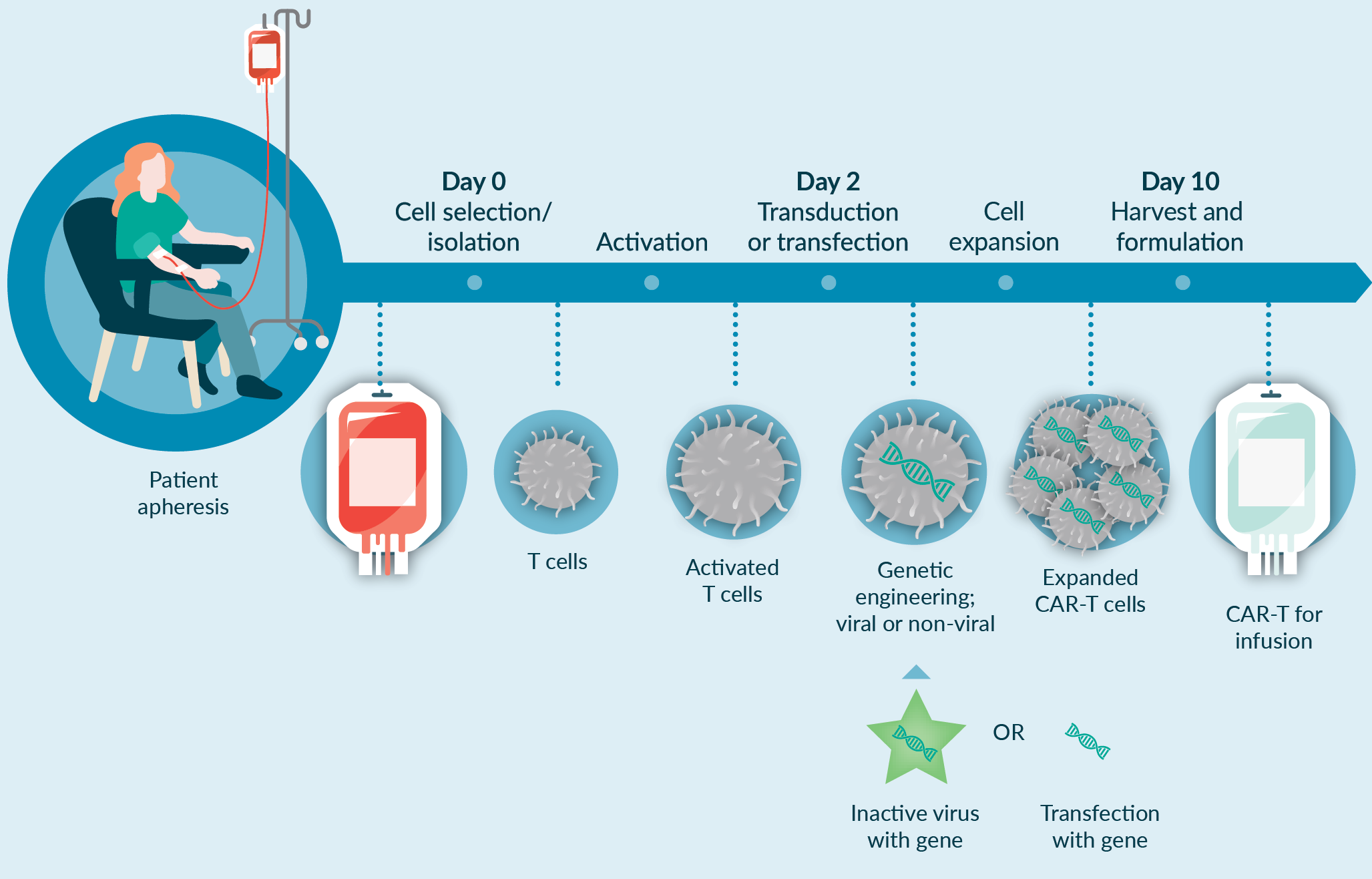
In order to optimize the unit operations (for example, cell isolation), one can run various scenarios and perform a wide range of optimization studies. These could include the evaluation of fresh versus frozen starting materials, the selection strategy (i.e. positive selection or negative selection), and also using different technologies and platforms during the isolation as highlighted in Figure 4
Depending on the approach we take from automation perspective, and technologies that are implemented in the process, the outcome of the process development and target criteria (e.g., for process recovery) could be different. The decision on the best process development strategy must be based on a phase appropriate, risk assessment, and evaluation of quality, safety, reproducibility, productivity and cost considerations.
Gene delivery is also another critical unit operation in the manufacturing of a CAR-T product. There are two main strategies for gene delivery: viral transduction or non-viral transfection. For each strategy, different process optimization and technologies (see Figure 5
Different gene modification technologies can be used for gene editing step. Lonza 4D-LV nucleofection system has been used in different CAR-T applications, and we have performed optimization and scale up studies moving from Lonza 4D Nuecleofector system to our larger scale (LV Nuceleofector) automated system while using different starting materials and gene delivery systems.
When using viral methods for gene delivery, the optimization of viral modification system may focus on initial titration study (to understand the transduction efficiency) and then performing titration study using multiple experimental conditions (for example, different combinations of media to enhance the efficiency of transduction).
Perhaps one of the most important steps in the development of a commercially viable manufacturing process is to pay specific attention to process analytics, in-process control and in-process monitoring, and implementation of appropriate control strategy in the process. Some of the suggested in-process controls for CAR-T applications include understanding the composition of the starting material (Box 1), monitoring changes in the composition following isolation, activation, before and after transduction or transfection (depending on the method of gene editing).
We strongly recommend defining the process control strategy and defining the input and output of each unit operation to ensure adequate control of the manufacturing process. Some proposed process controls for CAR-T cell therapies include:
- CD3, CD4 and CD8% of the starting material (apheresis);
- Post-selection T cell recovery;
- Post-selection cell purity (CD3%) and phenotype (memory subsets);
- Post-selection Impurities (RBC, platelets, tumor cells);
- Transduction, transfection or transposition efficiency;
- Monitoring for memory phenotype, activation state and exhaustion markers during expansion;
- Cell count and cell viability throughout culture and downstream processing;
- Process residual clearance.
In summary, to meet the growing demands as more cell and gene therapies move towards commercial-scale manufacture, it is important to implement proper process and assay development strategies, as well as using innovative technologies to establish a robust and reproducible automated closed system. Implementation of appropriate product release and characterization is going to be the key to maintaining the critical quality attributes of the final product. Finally, a phase appropriate development approach is needed to adjust the process and assay requirements for clinical and commercial applications.
| BOX 1 Characterization of starting material. |
|---|
|
Featuring the top 5 questions from our live webinar audience
You mentioned in your presentation a desire to move towards automating classically manual processes. But how do you go about automated density gradient processes for cell isolation?
At the moment there are a wide range of technologies available for automation of different unit operations including cell isolation step. First, it’s important to understand what is the final product quality requirement, or for each unit operations what would be the input and output quality attributes considering the process parameters. For example, if you are performing a study to evaluate processing of the starting material, then you should have a good understanding of the final volume, concentration, cellular compositions requirements. Then, using a wide range of technologies that are available, for instance Lovo Cell Processing, Elutra® Cell Separation System, SepaxTM Cell Processing Instrument, you would need to focus on the outputs and the quality attributes, incorporate appropriate technology (if possible through side-by-side comparability testing), and eventually optimize the process.
What is the largest size currently of 3D bioreactor for iPSC differentiation?
Currently most of the iPSC-based therapies are in early clinical stage, and based on my experience and understanding, the majority of applications may not need scale beyond 3 L bioreactors. We have actually worked with 50 L bioreactors for iPSC-based applications and I understand that as we move forward into clinical phases, larger scale bioreactor processes may be needed. So, we need to work based on a phase appropriate approach during the process development step and pay specific attention to bioreactor configurations, the scalability of a specific platform, scale, and clinical versus commercialization demands to properly choose the appropriate bioreactor technology and scale for each phase.
What do you see as the biggest roadblocks for cell & gene therapies to transfer from the lab to the market?
In my opinion, there are multiple challenges including GMP compliance (for instance, availability of GMP compliant materials) and understanding the critical steps of the process, and establishing a strong relationship between critical materials, critical process parameters, and critical quality attributes.
Another challenge is related to the assays that need to be implemented into the process. Depending on the applications, it is important to have a clear view of the type of assays and analytical methods that can be available to properly characterize starting materials, intermediate products, and also the final products.
Also, when these processes move to clinical manufacturing, harmonization and standardization of the approach for implementing new technologies and development of manufacturing processes need to be addressed.
In your opinion what do you think of the current state of assays we have available? Do you see this as being quite a significant bottleneck?
Absolutely! We know that the field of cell and gene therapy is growing, and also the need to develop new emerging assays is evolving. There are some characterization tools, analytical methods, that can be used, for instance flow cytometry based assays, molecular biology based assays, or cell-based assays. But the question we would need to answer is how we could incorporate emerging assays and technologies, for example whole genome sequencing, into manufacturing platforms.
At the moment, we proposed to use some of these assays in the characterization of starting materials, for instance iPSCs. If these methods could be qualified or validated, they could be used in the future quality control and release of the final product. This will allow improving product characterization and better control around the quality of the cell therapy products.
Can you speak about scaffold requirement in 3D bioreactors?
The use of scaffolds purely depends on the application. We know that the cellular microenvironment is important (in vitro) as we are trying to mimic the in vivo environment of the cells and maintain the therapeutic impact of the cell therapy products. Depending on the application, whether we want to minimize the immune rejection or whether we want to boost the therapeutic efficacy, we might need to use a scaffold and/or other biomaterial as a hybrid transplantation, along with the cell therapy product.
The type and implementation of scaffolds in 3D bioreactors will be largely dependent on the type of application, and how the cells are essentially inducing the therapeutic effect after the transplantation as well as strategies around control of immune rejection. The biomaterials field is significantly evolving, and it’s an important concept that will require attention in particular regarding the choice of materials, cell–cell interaction and cell-scaffold interaction, impact of the hydrodynamics of the bioreactors on the scaffolds and cells, cell format in the scaffolds (single cells vs cell aggregates), and the cellular therapeutic impact after transplantation into the patients.
Authorship & Conflict of Interest
Contributions: All named authors take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Acknowledgements: None.
Disclosure and potential conflicts of interest: There may be multiple patents associated with Lonza iPSC platform technology. The author declares that they have no other conflicts of interest.
Funding declaration: The author received no financial support for the research, authorship and/or publication of this article.
Article & copyright information
Copyright: Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0 which allows anyone to copy, distribute, and transmit the article provided it is properly attributed in the manner specified below. No commercial use without permission.
Attribution: Copyright © 2020 Baghbaderani BA. Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0.
Article source: This article is a transcript of a webinar entitled ‘Key steps to industrialize your process and de-risk your pathway to commercialization’, which can be found here.
Webinar published: Dec 16 2019; Publication date: May 4 2020.
References
1. Baghbaderani BA, Tian X, Neo BH et al. cGMP-Manufactured Human Induced Pluripotent Stem Cells Are Available for Pre-clinical and Clinical Applications. Stem Cell Reports 2015; 5(4): 647–59. CrossRef
2. Baghbaderani BA, Syama A, Sivapatham R et al. Detailed Characterization of Human Induced Pluripotent Stem Cells Manufactured for Therapeutic Applications. Stem Cell Rev Rep. 2016; 12(4): 394–420. CrossRef
3. Shafa M, Yang F, Fellner T, Rao MS, Baghbaderani BA. Human Induced Pluripotent Stem Cells Manufactured using a cGMP Compliant Process Differentiate into Clinically Relevant Cells from Three Germ-Layers. Frontiers Medicine 2018; 15(5) 69. doi: 10.3389/fmed.2018.00069. CrossRef
4. Shafa M, Panchalingam KM, Walsh T, Richardson T, Baghbaderani BA. Computational fluid dynamics modeling, a novel, and effective approach for developing scalable cell therapy manufacturing processes. Biotechnol. Bioeng. 2019; 116(12): 3228-3241. doi: 10.1002/bit.27159. CrossRef
Affiliations
Behnam Ahmadian Baghbaderani
Global Head of Process Development for Cell and Gene Technologies