
Scalable and efficient AAV production process with new Fibro chromatography technology
Cell & Gene Therapy Insights 2020; 6(8), 1249–1262
10.18609/cgti.2020.138
Production methods for adenovirus-associated virus (AAV) vectors have not kept up with the brisk pace of gene therapy development. To manufacture safe and efficacious clinical- grade virus, scalable and cost-effective production processes are needed. Towards this end, we present an efficient process for AAV production and scale-up in suspension cell culture through to purified bulk product. The process was developed by evaluating and optimizing each process step. A novel fiber technology, Fibro, addresses the downstream bottleneck at the capture step by overcoming the diffusional and flow limitations of purification using packed-bed chromatography. Also, a new analytical assay based on surface plasmon resonance was developed for AAV quantitation.
Introduction: challenges in improving AAV productivity & scalability
The utilization of AAV as a gene therapy vector has increased due to its relatively limited immunotoxicity and wide range of tissue tropism. To date, multiple AAV serotypes targeting different organs including brain, eye, lung liver, skeletal muscle, and heart have been discovered and characterized. Capsid proteins have been further modified to increase transduction, targeting specificity, and efficacy in vivo by methods that include:
- Directed evolution, which incorporates components of multiple AAV serotypes into the capsid;
- Random shuffling of capsid sequences to generate new novel capsids; and
- Adding aptamers to the surface.
AAV vectors are produced through the introduction of helper virus and AAV replication components into the production cell line. When these helper virus sequences are stressed, they trigger expression of both cellular factors critical in AAV production and activate the four different AAV Rep proteins (48, 52, 68, 78). These proteins are critical in AAV production, assisting in multiple functions including limiting replication of the packaging cell, expressing viral capsid proteins, and increasing production of the Cis DNA sequences. The replicated DNA sequences are then packaged into the AAV capsid and harvested from the cells and/or supernatant through the purification process. The ratios of these different viral and cellular helper proteins, as well as the AAV2 replication protein, help to dictate the overall number of particles packaged, the number of particles that contain DNA, and often the integrity or completeness of the genome that’s packaged within these capsids.
Currently, multiple methods are utilized to deliver each of these helper components and replication and capsid sequences into the cell. The most frequently used is plasmid transfection, where 2 to 3 different plasmids and DNA sequences are transfected into the cell. In some production systems, these sequences are packaged into recombinant viruses such as herpes, adenovirus, or baculovirus, which then transduce the production cell to initiate the production cascade. In addition, there are multiple cell types that are utilized, including HEK293, BHK, HeLa and insect Sf9 cells. Furthermore, both adherent and suspension platforms are frequently employed in this production process.
While research has continued to improve overall recombinant AAV production, the titers that are reported from the above systems are generally observed to be 5 to 10 times lower in productivity per cell than wild-type AAV, indicating that there are still learnings to be gleaned from the wild-type virus to help drive improvements in recombinant production systems.
Multiple strategies have been employed to improve both AAV production and product quality. Many of these begin with modifying the replication helper or capsid sequences.
One of the first modifications typically introduced to upstream bioprocesses is modifying the ratios of the plasmids or vector components that are going into the cell, with the goal of identifying the ideal amount needed for each specific serotype or Cis sequence to increase production of full capsids. Another strategy employed to improve control of the production cascade is modifying the amount of the replication proteins that are present, either through modifying the start codons, or using alternative constitutive or inducible promoters to better control which of those Rep proteins are expressed, and the timing of their expression within the cell. Furthermore, research focused on codon optimization of both the helper and AAV replication sequences is ongoing. Again, all of these strategies are working towards the end goal of increasing DNA replication within the production cell, improving packaging (or the number of full capsids that are produced in a system), and increasing the viral particle (VP) or capsid protein ratio in order to create a product that is more infectious.
Additional recent research has focused on modulating gene expression within the cell line to create a more favorable environment for AAV production. These studies typically involve evaluation of a panel of genes that are upregulated and downregulated before that gene regulation is correlated with improved AAV production and/or product quality. While there has been a wide range of genes reported to be associated with improved AAV production, a number that have garnered particular attention today have been linked to either membrane channel proteins, tumor suppression or regulation, controller transport at the nuclear envelope, or overall DNA replication. However, many of these studies have typically focused on improving just one production cell type or production system, without screening for applicability across multiple platforms.
The approaches described above are mainly aimed at creating a cellular environment and optimal viral gene expression that is more amenable to AAV production, and are typically employed at the raw material improvement stage by modifying either the plasmids or vector starting material in the initial cell banks. In AAV production, these materials are produced and characterized in a GMP-like environment. Following cell expansion with the producer cells that are optimal for AAV production, the next steps involve delivering these packaging components through either plasmid transfection or recombinant viral transduction into each cell. At this point, bioreactors can be further optimized, as can cell culture conditions (including the number of cells in culture media) to further improve AAV production. Depending upon the specific harvest strategy, virus is collected 3 to 7 days after initiating the production cascade. One of the remaining challenges in this field is ensuring the helper replication capsid and Cis sequences are introduced to each cell in an efficient and reproducible manner at the intended production scale.
Currently, the most commonly utilized method for AAV production is plasmid transfection due to its speed in initial material generation and relative flexibility in incorporating the sequence changes previously described. In transfection, DNA is mixed with a chemical that condenses it and creates a positively charged complex that can be endocytosed by the cell membrane. The overall amount of DNA, transfection reagent, and diluent components significantly impacts the quality of transfection complexes. These complexes grow over time as the mixture is incubated, eventually reaching a size where the complex is no longer easily taken up by the target cell. For these reasons, transfection is often cited as a difficult strategy to scale due to its relatively limited reaction time compared to the transfer rate into the production vessel.
Gravity draining of complexes has proven to be suboptimal at larger production scales due to the overall volumes required, as well as the time it takes to drain into the production bioreactor. Meanwhile, other groups have explored pumping a transfection mist, but this has not been associated with increased productivity due to potential damage to the transfection complex during pumping. Additionally, certain media components in the production vessel that are present at the time of transfection can decrease the efficiency of complex fusion and uptake by the target cell. However, despite these challenges, multiple groups have reported successful production using transfection-based methods at scales from 500 to 2,000 liters.
Besides the limited operating window and complex stability, the amount of plasmid required for AAV production has been cited as a limiting factor in the long-term feasibility of this method for delivering packaging sequences into production cells. While plasmid amounts are only in the 20 to 40 mg range in a 10-liter production, the 500 to 2,000 liter scales required for commercial products require grams of plasmid per production run. Additionally, variability can be observed in these methods due to the inherent difficulty in introducing equimolar amounts of the 2 to 3 plasmids used to the packaging cell line, which can then create a heterogeneous replication cascade across the entire production culture. Together, these issues can create limiting factors relating to both scalability and reproducibility of a transfection platform at scale.
An alternative method to transfection, helper viruses are utilized to deliver the replication capsid or Cis sequence into cells. Advantages commonly cited for these systems include the requirement for smaller amounts of helper virus compared to plasmids, improved stability of the helper viruses, and the fact that when they are placed into culture they demonstrate an improved ability to dispense through the entire production vessel and reach all of the cells. While helper viruses do potentially provide a more scalable method for delivering packaging components to cells, some production platforms still rely on the delivery of 2 to 3 recombinant helper viruses to each cell. This may result in a challenge similar to that faced by transfection: namely, difficulty in ensuring each cell receives an equimolar ratio expression of production sequences. Some baculovirus-based systems have utilized helper virus spread, where they infect with a low MOI (multiplicity of infection) and allow replication through the culture, further increasing the possibility of these systems being even more scalable. However, there are more stringent requirements for viral clearance studies, which are expected to evaluate the efficiency of the purification process in removing the input helper virus.
Finally, there is currently a strong interest in creating a true packaging cell line similar to those utilized in the protein therapeutics field.
To date, AAV vector developers have utilized various combinations of Cis, Rep, Cap, and/or helper sequences in the production cell. Integration of these components is particularly challenging for AAV production due to the need to carefully control the interaction of those genes involved in the production cascade, which places the onus on ensuring an optimal amount of each Rep protein as well as the adenovirus or other virus-based helper genes.
Multiple induction systems have been tested as a means of modifying which components are expressed and how much – an important step in both improving regulation of cytotoxicity that can come from the adenovirus replication gene, and ensuring an appropriate ratio of gene expression is gained during production.
Further challenges in developing a packaging cell line have been reported due to potential instability of the integrated ITR (inverted terminal repeats) due to its secondary structure. This instability can occur during cell expansion, during the banking process, or in cells that are expanded for production. In addition, some of the initial studies related to packaging cell lines that have been developed have reported a lower percentage of full capsids, or higher levels of genome truncation or package host cell DNA in the system. Despite these limitations, though, there are multiple groups utilizing this strategy, and considerable effort is being put into improving the platform to create a true packaging cell line in the AAV field.
Finally, it should be emphasized that careful evaluation of product quality should be paired with any bid to improve the upstream bioprocess. Modifications to the helper virus or production sequences have direct implications for overall productivity, but they also control the quality of the packaged genome or therapeutic sequence in terms of the amount of packaged impurity (e.g. host cell DNA, plasmid, or vector DNA sequences) in the infectivity of the protocol. By improving understanding of the virus biology and the production cascade involving helper, AAV, and cellular proteins, this interplay may be utilized to drive AAV systems closer to the cellular production capacity we currently see with wild-type AAV viruses.
Optimizing scalable AAV production: a case study
Upstream bioprocess
Cytiva recently developed a scalable upstream AAV bioprocess based upon triple plasmid transfection into HEK293T suspension cells. AAV2-GFP (containing a green fluorescent protein marker to aid in monitoring propagation) was the initial serotype selected.
The upstream process starts with the working cell bank, which is thawed before expansion in a number of shake flask steps. This is followed by virus production, which is carried out in the Xcellerex™ XDR-10 stirred tank single-use bioreactor, scalable to 2,000 liters.
Optimization of the cell line involved adapting the HEK293T cell line to suspension culture. (It may be noted that parallel development of a HEK293 cell-based process is underway, in light of recent debate around the large T antigen and its possible regulatory implications). A chemically defined cell culture medium was used to avoid the regulatory complications associated with animal-derived components such as serum. Next, the cell density and expansion procedure was optimized before creation of the cell bank.
Design of Experiments (DoE) approaches were utilized in optimizing the transfection procedure. A number of different parameters were explored, including cell density, volume of transfection, various different plasmids and the concentrations and ratios between them, PEI (polyethylenimine)/plasmid ratio, incubation time prior to entering the bioreactor, temperature, different supplements (including sodium butyrate), and time of harvest post-transfection.
Figure 1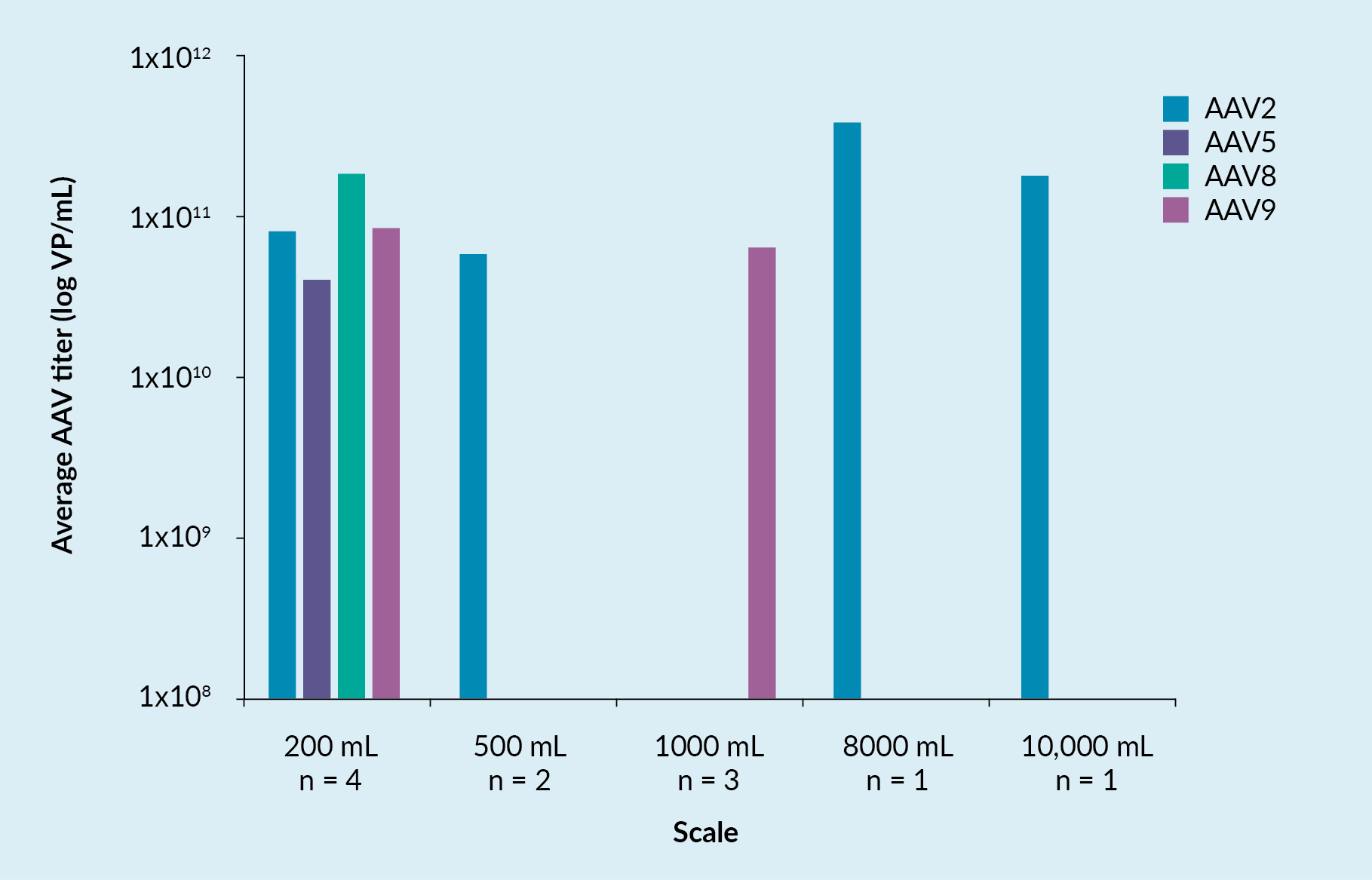
Downstream bioprocess
Figure 2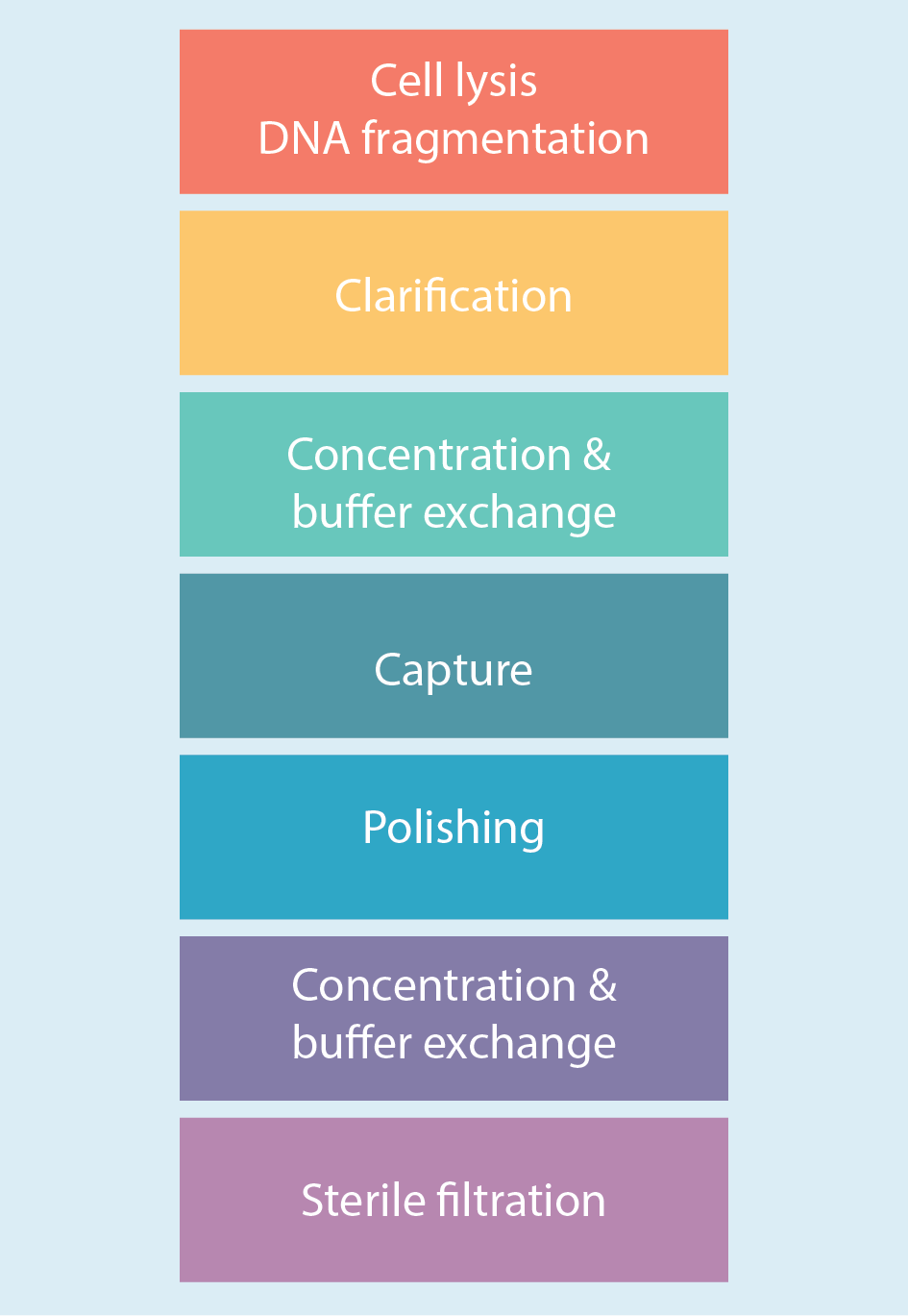
Evaluation and optimization of the downstream purification steps naturally began at small scale and with a focus on the cell lysis and DNA fragmentation step. A combination of 0.5% Tween™ 20 (to lyse the cells) and 40 U/mL Denarase™ (to digest DNA) was employed. This step took place in the bioreactor at 37°C with a mixing time of 4 hours.
A normal flow filtration step followed using ULTA™ capsules with a range of different cutoffs (5 µm + 2 µm + 0.6/0.2 µm HC) and flow rates (30 to 50 LMH). Recovery from this step was approximately 75% to 80%, with the usual slight variability between runs observed and the inevitable loss of some virus (e.g. through it sticking to cell debris, etc.)
A concentration and buffer exchange step followed, which was done by tangential flow filtration using hollow fiber filters. Figure 3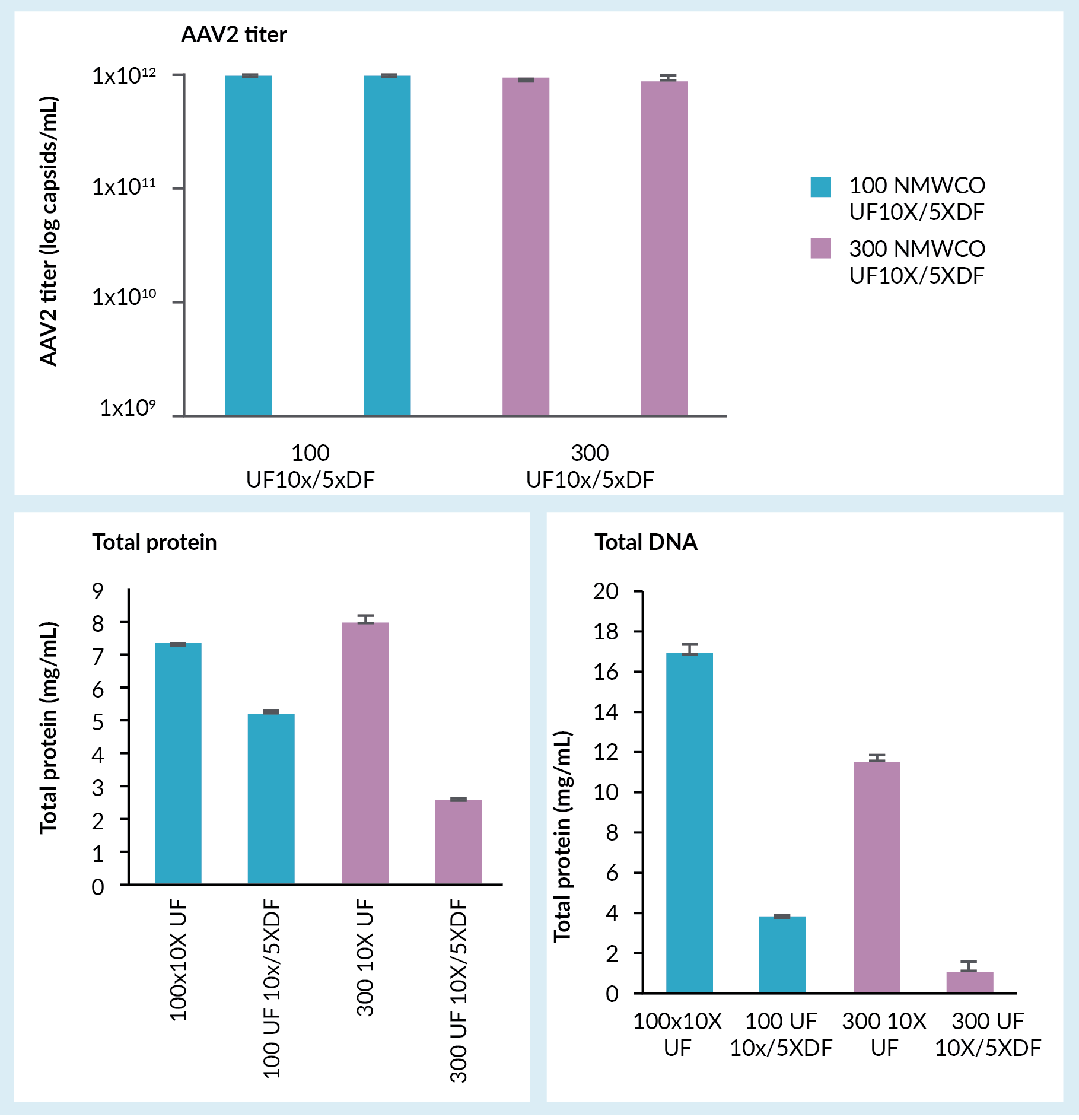
The capture step involved affinity chromatography utilizing Cytiva’s Capto AVB affinity resin. Figure 4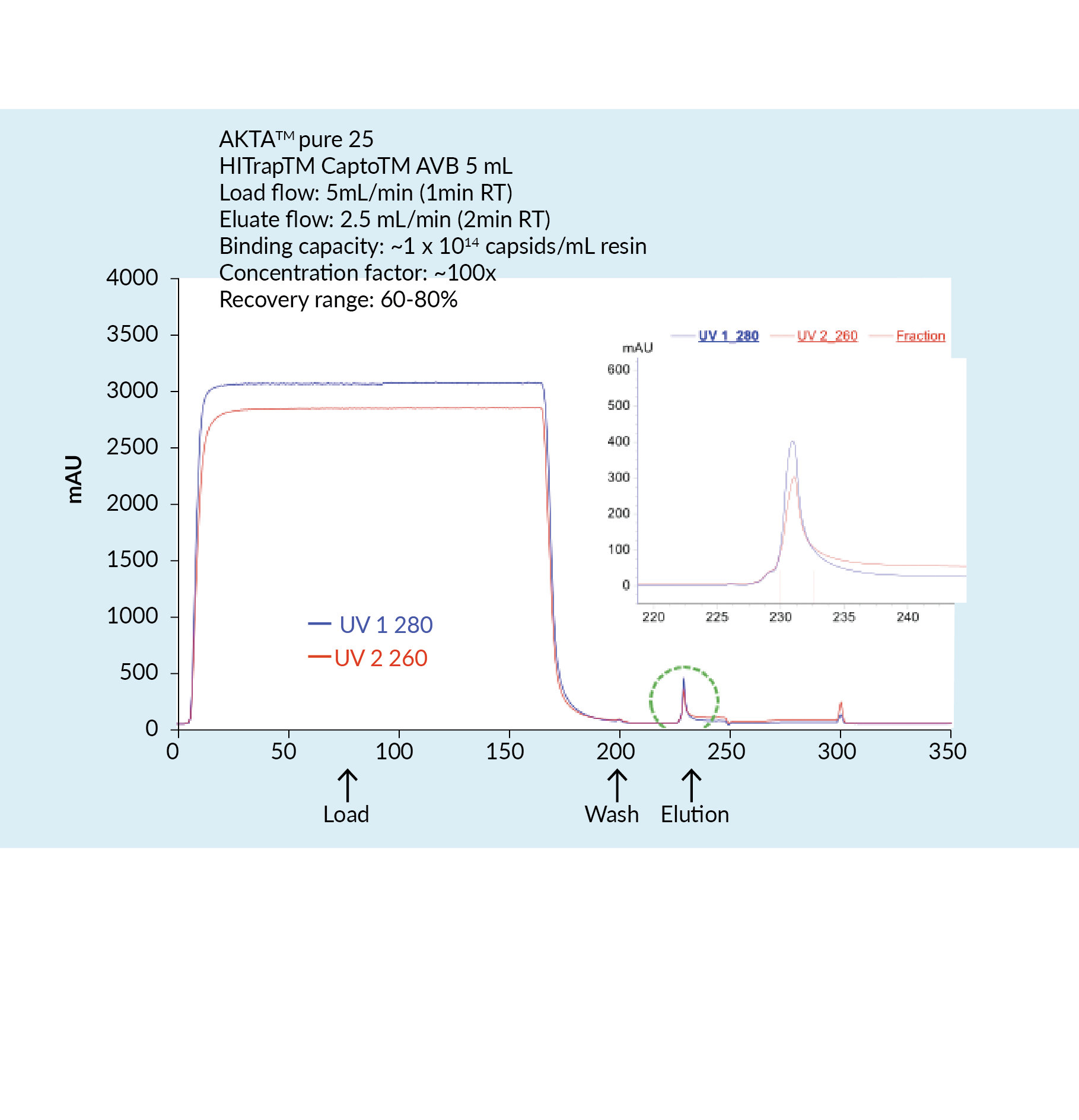
This proved to be a very efficient purification method with high impurity reduction achieved from a single chromatography step. Figure 5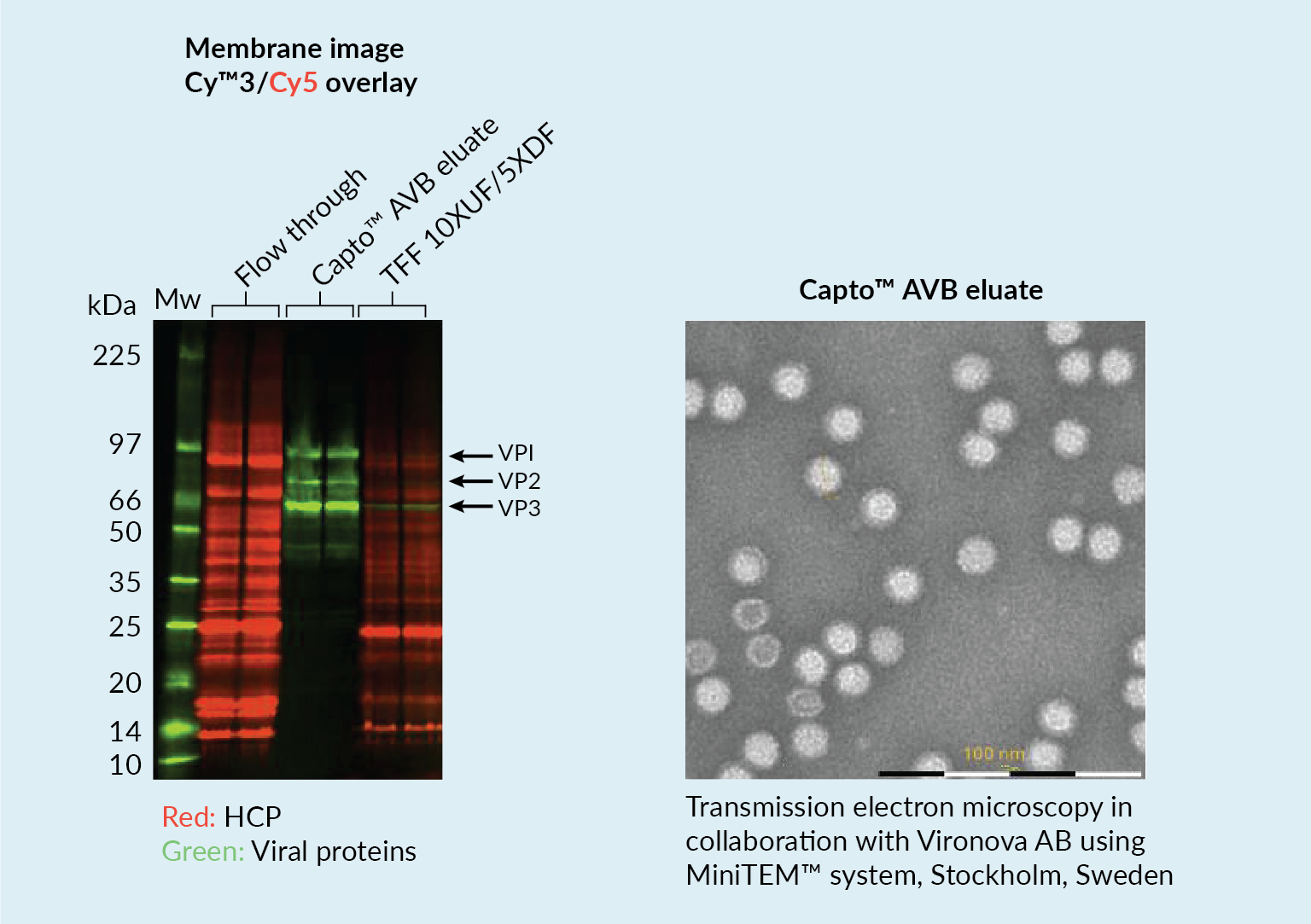
While efficiency is of course important, it is equally critical to build robustness into the process. With this in mind, ongoing development work is focusing on aspects such as empty-full capsid separation and polishing any remaining impurities. The particular strategy under development involves using high resolution anion exchange resins (for example, Capto™ Q ImpRes) although a number of other alternatives are currently being explored.
Analytics
Analytics are critical for bioprocess success, but they are also very time-consuming – an all-too-familiar issue for anyone who has worked in virus production.
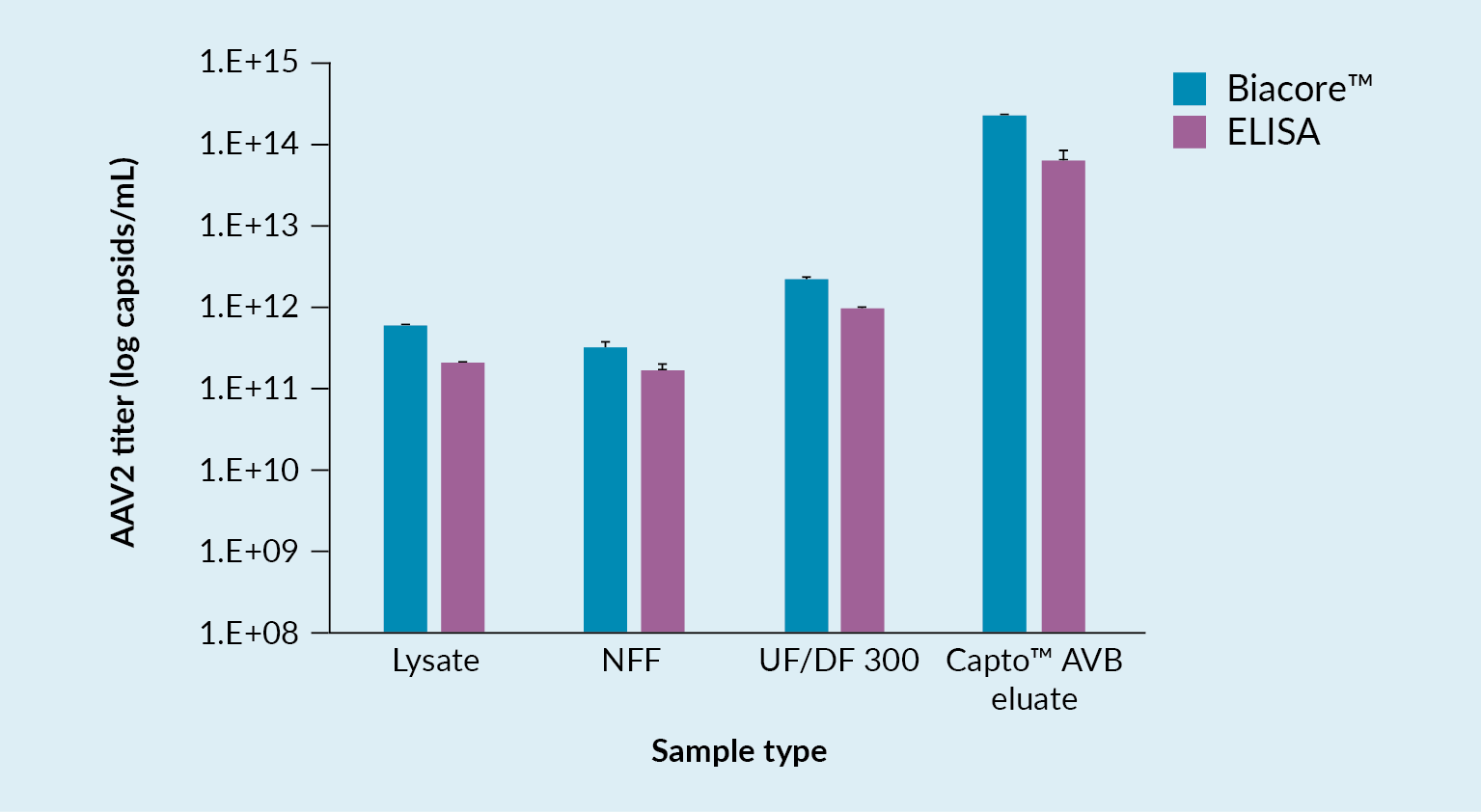
Box 1 lists the analytical tools that were utilized to follow the above process, including a novel technology based on Biacore™ SPR (Surface Plasmon Resonance) technology, which was developed for determination of virus titer. Assays used included infectious titer and total virus titer assays and, of course, a number of assays for host cell impurities and vector characterization.
BOX 1Virus infectious titer
Virus titer
Host cell impurities
Characterization
|
Challenges faced with these methods include the fact that some are lacking low enough limits of detection, especially when used with early-stage samples from the harvest and the NFF samples. Additionally, they can sometimes be affected by detergents or buffer components, their accuracy depends to a large extent on the sample impurity level, and there is a lot of assay variation.
In a bid to overcome some of these challenges and limitations, a new quantification assay for AAV2 was developed using the Biacore™ T200 instrument. This instrument carries a sensor chip to which is bound an anti-AAV2 antibody. The antibody is immobilized by amine coupling. As material flows over the chip, interaction between the AAV and the antibody may be detected. Figure 6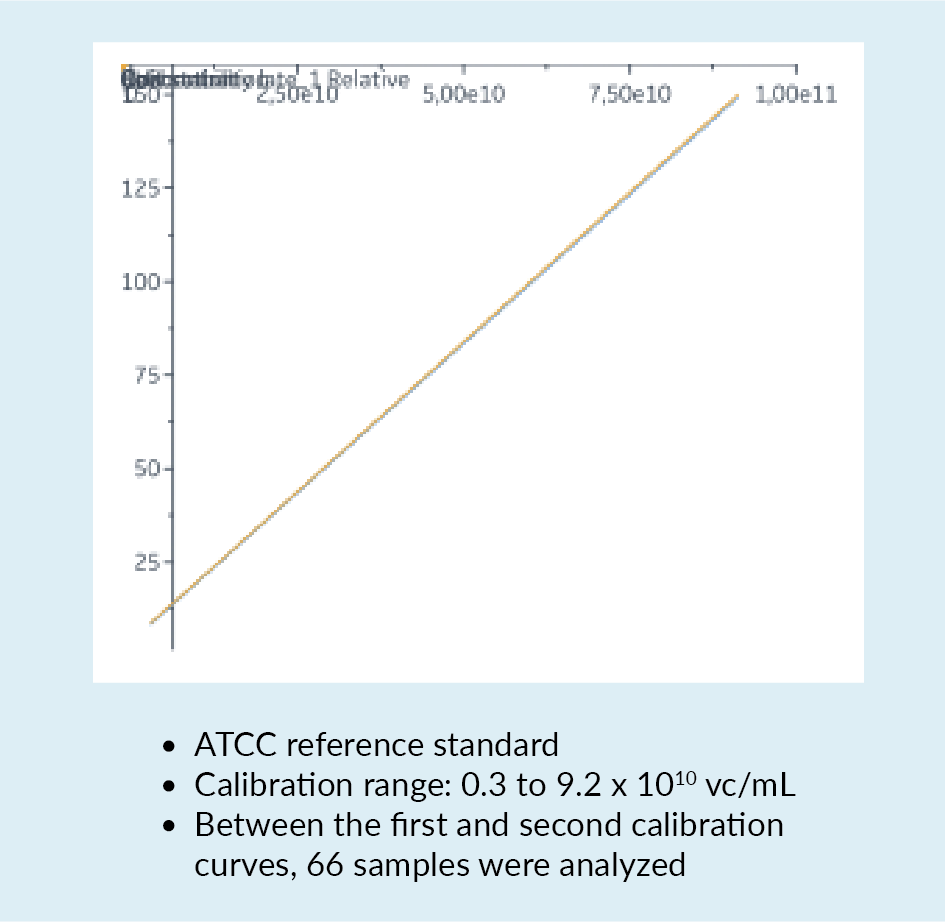
Fibro chromatography in AAV processing
Fibro is a novel, single-use chromatography tool with the potential to alleviate several of the current challenges in AAV downstream processing, including those related to speed and process efficiency. However, it may also positively impact other, more general manufacturing pain points, such as speed to market, scalability, and Cost of Goods.
The Fibro technology is mainly applicable to the capture step of AAV downstream bioprocessing. Currently, the capture step typically provides good recovery, although it can be improved, but it is also a relatively slow process step.
Fibro technology enables one to address both the capture and the prior concentration step.
Concentration typically has a recovery of approximately 80% and like capture, it is widely considered to be relatively slow and time-consuming. The reason for introducing this step is to concentrate the feed material and to minimize the loading time in the subsequent capture step. In some cases, a buffer exchange is also done in this concentration step, usually by TFF.
Capture steps using affinity resins are usually associated with very long loading times. This is due to the fact that AAV titers are relatively low, and the flow properties of the resins mean that extended residence times are required – usually 1 to 3 minutes. Consequently, large volumes of feed material may take a great deal of time to load. Indeed, columns are sometimes oversized to minimize time spent on this loading phase. Recovery in the capture step is negatively impacted by this long process time as is the low pH elution, which is usually used for affinity ligands.
Fibro is an electrospun cellulose material that has relatively large pore size, allowing the ligands that are immobilized on this format to be accessible directly, without any need for diffusion. Therefore, residence times of only a few seconds are needed.
There are alternatives available on the market – bead resin, for example. Bead resin has a much larger surface area, but the majority of this surface area is not accessible by the relatively large AAV. Other materials are not dependent on diffusion. However, these do not have the same surface area, and they have a different structure without the same, even pore size distribution that Fibro features. Fibro has a large surface area and also has a high binding capacity, which is key for AAV.
In the process scenario demonstrated in Figure 8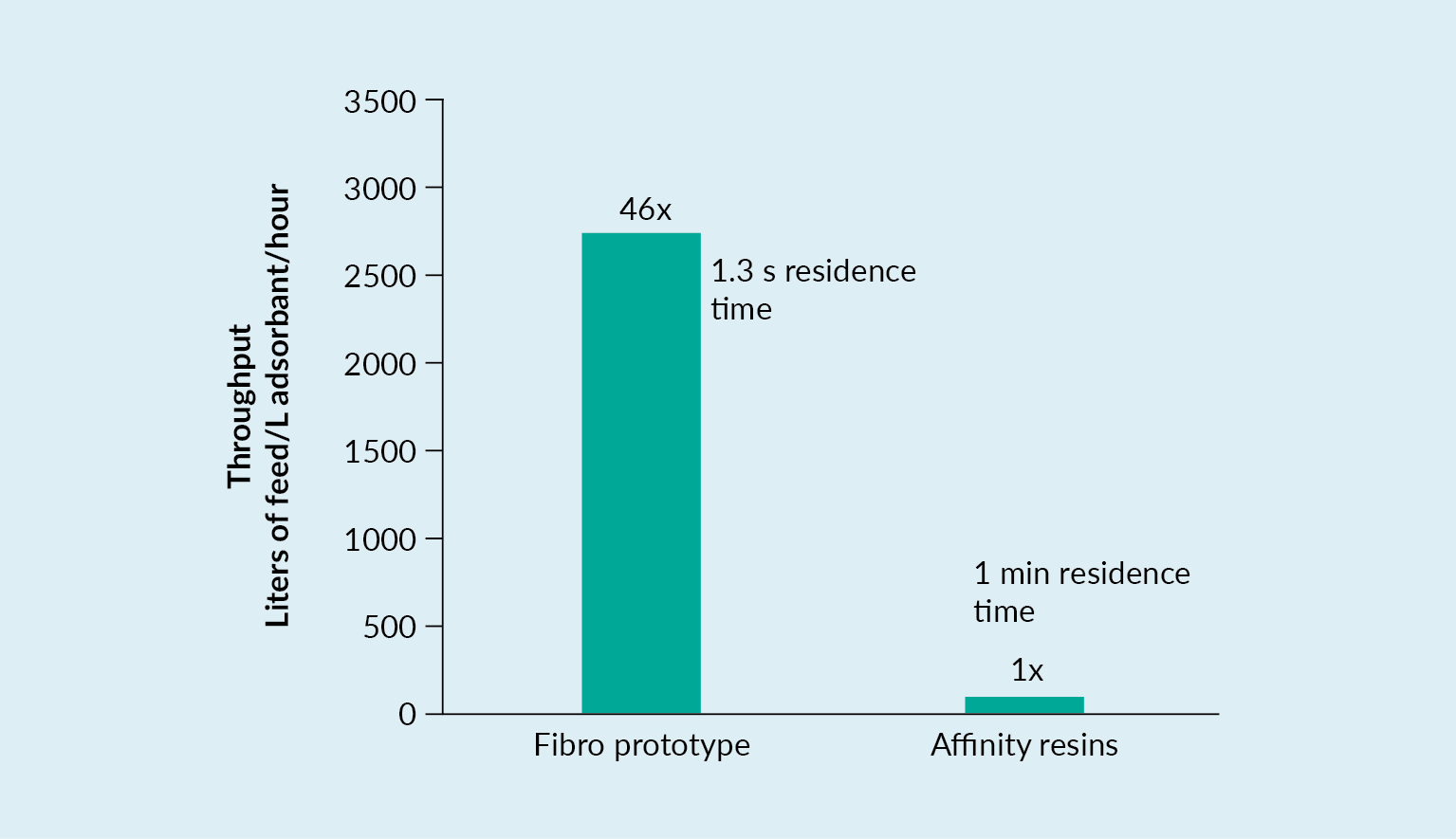
Table 1 shows different residence times and different flows. The capacity is dependent on the flow, but it will remain high even at a short residence time of 1 to 2 seconds, only starting to decrease at approximately 0.5 seconds. The right-hand column of the table shows the corresponding time to process 2,500 MV (membrane volumes), which further demonstrates the speed with which one can process clarified feed material even without a preconcentration step.
| Table 1 | |||
| Residence time (s) | Flow (MV/min) | Capacity (capsids/mL) | Approx. process time for 2500 MV |
| 0.5 | 120 | 6.1 x 1013 | 25 min |
| 1 | 60 | 2.0 x 1014 | 50 min |
| 2 | 30 | 3.4 x 1014 | 90 min |
In order to provide context in terms of the quantity of Fibro material required to purify a large-scale run: 1 liter of feed material on a 400 microliter Fibro unit corresponds to 500 liters of feed material needing a few hundred milliliters of Fibro material.
A number of different units are being developed that range in size from the lab-scale 400 microliter HiTrap™ unit up to the 2.4-liter Large Fibro unit. The Medium Fibro unit (160 mL) is capable of processing 500 liters of feed material in a single run. Each unit is compatible with corresponding hardware that is suitable to cope with the flows involved.
In summary, Fibro offers a number of advantages for the AAV capture step, including speed, capacity, recovery, and the efficiency and convenience of a single-use format.
The fast flow negates the requirement for a prior sample concentration step. Rapid cycle times positively impact maintenance of both virus integrity and infectivity. Rapid processing is also positive for the recovery, and by avoiding the need for a TFF step, any losses associated with that step are removed. Fibro units are prepacked, with a simple setup in the manufacturing facility, and offer short process development timelines due to the speed of every cycle.
Note that Fibro units for AAV are still under development.
Affinity vs multimodal ligands for AAV capture
Affinity ligands are now well established on the market for most AAV serotypes. A key strength is their prowess at impurity removal, due to their high specificity in binding the target AAV.
Affinity ligands do have drawbacks. These include the challenging elution conditions with low pH – a particularly important consideration for runs with resins because of lengthy timeframes involved. Additionally, those affinity ligands currently available on the market do not discriminate between full and empty particles. The elution conditions usually raise the risk of the AAV sample aggregating and finally, protein-based affinity ligands have limited cleanability. However, efficient impurity removal with affinity ligands may in some cases be considered unnecessary because one may simply introduce a subsequent full/empty separation step, which will also remove many impurities.
Multimodal ligands are not as effective as affinity ligands at impurity removal, but they do hold the advantage of very mild elution conditions. There is an opportunity to enrich full AAV, depending on how one runs the elution and collects the elution peak. Multimodal ligands are acceptable with high conductivity during binding, meaning that no buffer exchange is required prior to loading. They minimize the risk of aggregation. Being synthetic, they may also be cleaned under very harsh conditions. Finally, they have broad cell type coverage.
There also exists an opportunity to circumvent multimodal ligands’ limited impurity removal capabilities, even if one wishes to avoid a subsequent full/empty separation step: residual HCP and DNA can be removed through the addition of a gentle flow through Capto™ Core 400, following a capture step with a multimodal ligand.
Conclusion
Significant improvements have been made to vector productivity and genome packaging by modifying both viral and cellular sequences to better support AAV production. However, with the large range of production systems still used to manufacture AAV, these improvements may need to be reoptimized with each platform to better control the production cascade. Further challenges remain in the scalability of these methods.
As these platforms continue to improve, early assessments of product quality will increase the field’s understanding of the link between viral gene expression, replication, and improving AAV platforms.
The optimization of processes and the continued emergence of improved upstream and downstream steps, tools, and assays such as those described above, will play a key role in evolving traditional AAV virus production into fit-for-purpose commercial gene therapy manufacture.
Affiliations
Dr Laura Adamson-Small
Upstream and Analytical Development Lead, UCB Biosciences
Dr Mats Lundgren
Customer Applications Director, Cytiva
Dr Peter Guterstam
Product Manager, Next Generation Resins & Technologies, Cytiva
![]()

Authorship & Conflict of Interest
Contributions: All named authors take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Acknowledgements: None.
Disclosure and potential conflicts of interest: The authors declare that they have no conflicts of interest.
Funding declaration: The authors received no financial support for the research, authorship and/or publication of this article.
Article & copyright information
Copyright: Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0 which allows anyone to copy, distribute, and transmit the article provided it is properly attributed in the manner specified below. No commercial use without permission.
Attribution: Copyright © 2020 Name INTL. Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0.
Article source: This article is based on the transcript of a webinar held on the 30th July 2020, which can be viewed here.
Publication date: Oct 01 2020.