Process development and scale-up of pluripotent stem cell manufacturing
Cell & Gene Therapy Insights 2020; 6(9), 1277–1298
10.18609/cgti.2020.141
Human embryonic stem cells (hESCs) and induced pluripotent stem cells (hiPSCs), together human pluripotent stem cells (PSCs), have tremendous potential for production of cellular therapies for regenerative medicine. The final therapeutic dose of differentiated PSCs varies according to application; however, most indications have production requirements that cannot be met by traditional static tissue culture methods. Stirred-tank reactor expansion of PSCs represents a scalable solution to meet this developing demand. Here we present the process development of a 10 L single-use stirred tank bioreactor platform with the ability to generate > 1010 PSCs per production run and its applicability across both hESC and hiPSC cell lines. Manufacturing advancements are also presented. High-density seed bank inoculation of the process is demonstrated to decouple the adherent tissue culture requirement and normalize the cellular process input, as well as shorten the time requirement of the suspension seed train. Process correlations of pH and dissolved oxygen to viable cell densities are presented to remove or minimize the sampling requirements during expansion. Automated downstream processing, with volume reduction and washing, is demonstrated with 80–90% cell recovery and >94% viability. Together, these works represent seminal steps in the development of a controlled, automated and defined manufacturing process for PSCs.
Introduction
Human pluripotent stem cells (PSCs) have the capacity to become the source of many cell therapies in the regenerative medicine space due to their unique characteristics of unlimited self-renewal and differentiation potential to repair and regenerate diseased or damaged cells, organs and tissues [22]Reubinoff BE, Pera MF, Fong CY, Trounson A, Bongso A. Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nat. Biotechnol. 2000; 18: 399–404., [25]Takahash, K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007; 131: 861–872., [26]Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science 1998; 282: 1145–1147.. The possible applications for these cells are extensive, and their therapeutic potential is being investigated for use in a wide array of clinical areas, including those associated with high morbidity and mortality rates on a global scale, such as heart failure [8]Hartman ME, Da DF, Laflamme MA Human pluripotent stem cells: Prospects and challenges as a source of cardiomyocytes for in vitro modeling and cell-based cardiac repair. Adv. Drug Deliv. 2016; Rev 96: 3–17., [13]Van Laake LW, Hassink R, Doevendans PA, Mummery C. Heart repair and stem cells. J. Physiol. 2006; 577, 467–478., Parkinson’s disease [7]Hargus G, Cooper O, Deleidi M, et al. Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proc. Natl. Acad. Sci. 2010; 107, 15921–15926. and type I diabetes [6]D’Amour KA, Bang AG, Eliazer S et al. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat. Biotechnol. 2006; 24, 1392–1401., [10]Kroon E, Martinson LA, Kadoya K et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 2008; 26: 443–452..
Currently, PSC-based clinical trials have been initiated for therapies targeting age-related macular degeneration of the retina, spinal cord injury, type I diabetes and myocardial infarction, among others [27]Trounson A, DeWitt ND. Pluripotent stem cells progressing to the clinic. Nat. Rev. Mol. Cell Biol. 2016; 17, 194–200.Trounson A, DeWitt ND. Pluripotent stem cells progressing to the clinic. Nat. Rev. Mol. Cell Biol. 2016; 17, 194–200., [28]Trounson A, Thakar RG, Lomax G, Gibbons D. (2011). Clinical trials for stem cell therapies. BMC Med. 2011; 9: 52.Trounson A, Thakar RG, Lomax G, Gibbons D. (2011). Clinical trials for stem cell therapies. BMC Med. 2011; 9: 52.. Yet, despite a large and ever-growing body of preclinical research supporting the regenerative potential of PSCs, most clinical trials remain small-scale and run through academic groups [14]Li MD, Atkins H, Bubela T. The global landscape of stem cell clinical trials. Regen. Med. 2014; 9: 27–39.Li MD, Atkins H, Bubela T. The global landscape of stem cell clinical trials. Regen. Med. 2014; 9: 27–39.. While clinical trials using human embryonic stem cells (hESC) to treat ocular degenerative diseases have proceeded with cell dosage numbers in the 1 x 105 cell range, it is estimated that most proposed PSC-based cell therapies, including those targeting some of the leading causes of morbidity and mortality—heart disease, type I diabetes and Parkinson’s disease for example—could require as many as 2 x 109 PSCs per patient dose [9]Kempf H, Kropp C, Olmer R. Cardiac differentiation of human pluripotent stem cells in scalable suspension culture. Nat. Protoc. 2015; 10: 1345–1361., [29]Zweigerdt R. Large scale production of stem cells and their derivatives. Adv. Biochem. Eng. Biotechnol. 2006; 114: 201–235.Zweigerdt R. Large scale production of stem cells and their derivatives. Adv. Biochem. Eng. Biotechnol. 2006; 114: 201–235.. Given the large cell numbers required, traditional strategies of PSC propagation, which rely on 2D adherent culture, are neither practical nor cost efficient enough to meet the cell numbers required for PSC-based therapies.
To enable translation of these therapies into the clinic and to attract the interest of business partners willing to fund the emerging cell therapy industry, the field must meet the challenge of large-scale production, manufacturing and commercialization of these cells at clinically relevant titers [14]Li MD, Atkins H, Bubela T. The global landscape of stem cell clinical trials. Regen. Med. 2014; 9: 27–39.Li MD, Atkins H, Bubela T. The global landscape of stem cell clinical trials. Regen. Med. 2014; 9: 27–39.. As such, one of the limitations facing the implementation of these powerful cells as a tool to fight disease is the development and standardization of a robust, scalable and cost-effective production solution able to meet good manufacturing practices (GMP) and regulatory approval guidelines [27]Trounson A, DeWitt ND. Pluripotent stem cells progressing to the clinic. Nat. Rev. Mol. Cell Biol. 2016; 17, 194–200.Trounson A, DeWitt ND. Pluripotent stem cells progressing to the clinic. Nat. Rev. Mol. Cell Biol. 2016; 17, 194–200., [28]Trounson A, Thakar RG, Lomax G, Gibbons D. (2011). Clinical trials for stem cell therapies. BMC Med. 2011; 9: 52.Trounson A, Thakar RG, Lomax G, Gibbons D. (2011). Clinical trials for stem cell therapies. BMC Med. 2011; 9: 52..
Historically, cell scale-up and manufacturing efforts have employed stirred tank reactors (STRs) to maximize cell expansion by ensuring optimal mixing of gasses, nutrients and growth factors, while allowing for tight control and monitoring of key culture parameters and minimizing costs associated with production [3]Bareither R, Pollard D A review of advanced small-scale parallel bioreactor technology for accelerated process development: current state and future need. Biotechnol. Prog. 2011; 27, 2–14., [29]Zweigerdt R. Large scale production of stem cells and their derivatives. Adv. Biochem. Eng. Biotechnol. 2006; 114: 201–235.Zweigerdt R. Large scale production of stem cells and their derivatives. Adv. Biochem. Eng. Biotechnol. 2006; 114: 201–235.. Based on previous work done with murine embryonic stem cells (ESCs), studies have shown that human PSCs are amenable to suspension culture under defined and growth factor-supported conditions, maintaining key phenotypic markers, pluripotency and proliferative abilities required for stem cell identity. These studies saw cell concentrations in the 0.5–2 x 106 cells/mL range in spinner or shake flasks at the 5–25 mL scale, through cell aggregate formation [1]Amit M, Chebath J, Margulets V, Laevsky I et al. (2010). Suspension Culture of Undifferentiated Human Embryonic and Induced Pluripotent Stem Cells. Stem Cell Rev Rep. 2010; 6: 248–259., [2]Amit M, Laevsky I, Miropolsky Y et al. (2011). Dynamic suspension culture for scalable expansion of undifferentiated human pluripotent stem cells. Nat. Protoc. 2011; 6: 57, [17]Olmer R, Haase A, Merkert S, et al. Long term expansion of undifferentiated human iPS and ES cells in suspension culture using a defined medium. Stem Cell Res. 2010; 5: 51–64., [19]Phillips BW, Horne R, Lay TS, Rust WL, Teck TT, Crook JM. Attachment and growth of human embryonic stem cells on microcarriers. J. Biotechnol. 2008; 138: 24–32.. Since this important early work, the process has been optimized by moving PSC cultures into stirred tank reactor vessels with pH and dissolved oxygen (DO) control abilities, allowing for scale-up and the production of up to 2 x 108 cells in a 100 mL volume (2 x 106 cell/mL) [18]Olmer R, Lange A, Selzer S, et al. Suspension culture of human pluripotent stem cells in controlled, stirred bioreactors. Tissue Eng. Part C Methods 2012; 18: 772–784.. The addition of an effective perfusion system with cell retention capabilities compatible with these cells allowed for even higher cell concentrations, in the 3 x 106 cell/mL range at the same volume scale [11]Kropp C, Kempf H, Halloin C, et al. Impact of Feeding Strategies on the Scalable Expansion of Human Pluripotent Stem Cells in Single-Use Stirred Tank Bioreactors. Stem Cells Transl. Med. 2016; 5: 1289–1301.Kropp C, Kempf H, Halloin C, et al. Impact of Feeding Strategies on the Scalable Expansion of Human Pluripotent Stem Cells in Single-Use Stirred Tank Bioreactors. Stem Cells Transl. Med. 2016; 5: 1289–1301.Kropp C, Kempf H, Halloin C, et al. Impact of Feeding Strategies on the Scalable Expansion of Human Pluripotent Stem Cells in Single-Use Stirred Tank Bioreactors. Stem Cells Transl. Med. 2016; 5: 1289–1301..
While these cell titers show great promise in the scalability of pluripotent cultures, final scale-up of this process will entail culture of these cells at much larger volumes, requiring new optimization and control of parameters to overcome large scale-specific challenges such as oxygen and nutrient transport, homogeneity of cell aggregate sizing and shear sensitivity [11]Kropp C, Kempf H, Halloin C, et al. Impact of Feeding Strategies on the Scalable Expansion of Human Pluripotent Stem Cells in Single-Use Stirred Tank Bioreactors. Stem Cells Transl. Med. 2016; 5: 1289–1301.Kropp C, Kempf H, Halloin C, et al. Impact of Feeding Strategies on the Scalable Expansion of Human Pluripotent Stem Cells in Single-Use Stirred Tank Bioreactors. Stem Cells Transl. Med. 2016; 5: 1289–1301.Kropp C, Kempf H, Halloin C, et al. Impact of Feeding Strategies on the Scalable Expansion of Human Pluripotent Stem Cells in Single-Use Stirred Tank Bioreactors. Stem Cells Transl. Med. 2016; 5: 1289–1301..
Recently, Kwok and colleagues have demonstrated a suspension culture for PSC propagation using single-use bioreactors, able to produce 2 x 109 PSCs with full pluripotency retention, for the first time establishing the scalability of PSC culture to clinically relevant cell numbers [12]Kwok CK, Ueda Y, Kadari A et al. Scalable stirred suspension culture for the generation of billions of human induced pluripotent stem cells using single-use bioreactors. J. Tissue Eng. Regen. Med. 2018;. Yet, while this study demonstrated the production of cell numbers sufficient to meet the needs of a single patient dose, new research is driving the field towards the possibility of allogeneic PSC therapies, introducing the need for a PSC culture process able to produce enough cells to treat hundreds or even thousands of patients at a manufacturing scale [4]Carpenter M.K, Rao MS Concise Review: Making and Using Clinically Compliant Pluripotent Stem Cell Lines. Stem Cells Transl. Med. 2015; 4, 381–388., [23]Sackett SD, Brown ME, Tremmel DM, Ellis T, Burlingham WJ, Odorico JS. Modulation of Human Allogeneic and Syngeneic Pluripotent Stem Cells and Immunological Implications for Transplantation. Transplant. Rev. Orlando Fla 2016; 30: 61–70.. As such, the field still requires process intensification and optimization to (1) continue the scale-up of these PSC cultures towards clinical manufacturing volumes and (2) achieve large-scale PSC manufacture that meets regulatory specifications and is compatible with GMP manufacture and clinical use.
In the present study, we demonstrate a reproducible protocol for the scaled-up expansion of pluripotent stem cells in 10 L single-use stirred tank bioreactors. This could lend itself to further scale-up, as the platform has a demonstrated, incremental path to 2000 L in traditional bioprocess applications (i.e. monoclonal antibody production). Here, we detail the process development approach taken to develop this protocol and scale these cultures to produce >1010 cells per batch, providing clinically and industrially relevant quantities towards their use in therapeutic regenerative medicine applications. With attention to future GMP compliance and regulatory considerations for clinical manufacture, our use of detailed monitoring of in-process parameters and closed processing protocols allowed for minimal in-process sampling, which will lend itself to future automation and reduced contamination risk. Finally, we have demonstrated a downstream processing approach for volume reduction and washing of the final PSC product and present cases where this product was successfully used either directly, or as a cryopreserved high-density seed bank (HDSB), as inoculum for further stirred tank bioreactor expansion and a suspension-based downstream differentiation.
The high-density cryopreservation of PSCs provides a consistent inoculum to the manufacturing process. These HDSBs remove the need for upstream adherent tissue culture, normalize the manufacturing process input and reduce the overall time required to produce a 10 L batch. Direct inoculation of suspension-based differentiation serves as a game-changer in process development and eventual clinical manufacturing by decoupling the expansion and differentiation processes.
Materials and Methods
Maintenance of human PSCs in 2D culture
ESI-017 (hESCs, ESI BIO, Alameda, CA, USA) and NCRM1 (iPSCs, National Institutes of Health, Bethesda, Maryland, USA) were cultured at 37°C, 5% (v/v) CO2 and maintained in mTeSR™1 medium (STEMCELL Technologies, Vancouver, BC, Canada) on Matrigel® - coated (Corning, New York, NY, USA) flasks. Cells were cultured in T-75 flasks (BD Falcon, Fisher Scientific, Carlsbad, CA, USA), in a 12 mL volume. The medium was refreshed daily. When reaching approximately 75% confluence, cells were washed with PBS (Gibco, Grand Island, NY, USA) and dissociated in Gentle Cell Dissociation Reagent (GCDR, STEMCELL Technologies) for 6–8 min. Cell colonies were then detached by scraping with a cell scraper. A uniform suspension of cell aggregates was obtained by carefully pipetting the mixture up and down. The cell aggregate suspension was plated at a density of 20,000 cells/cm2 (or 1.5 x 106 cells/flask) on the pre-coated flasks with Matrigel and maintained in mTeSR1.
Expansion of PSCs in stirred tank bioreactors
PSCs were cultured adherently on Matrigel-coated flasks as described above. When they reached ~75% confluence in flasks, cells were harvested by treatment with TrypLE™ Select 1X (Thermo Fisher Scientific) for 5 min at 37°C. Dissociated cells were quenched with DMEM/F12 (Thermo Fisher Scientific) and centrifuged at 300 x g for 5 min at room temperature. The cell pellet was gently resuspended in 40 mL mTeSR1. The cell suspension was strained through a 70 µm cell strainer (Thermo Fisher Scientific) to remove undissociated clumps. Cells were counted and viability was determined using a NC-200™ NucleoCounter™ (Chemometec, Allerod, Denmark) and inoculated at a cell density of 2.5 x 105 cells/mL into a 250 mL DASbox™ vessel (Eppendorf, Hamburg, Germany). Cells were grown in mTeSR1, supplemented with 10µM rho-associated protein kinase inhibitor Y27632 (Tocris Bioscience, Bristol, United Kingdom), for the first 24 hours of batch suspension culture. The bioreactor was maintained at 37°C, pH 7.2, 50% DO, stirred at the optimized dynamic agitation profile (specified in results), and supplied with sterile-filtered air and CO2 via an overlay. Perfusion was initiated 24 hours post-inoculation with either a fixed perfusion rate or enhanced perfusion rate (see Results section for detailed perfusion protocols), via a stock micro-sparger used in reverse orientation. Process control and continuous data acquisition were executed by DASGIP™ software (Eppendorf). Daily sampling included cell counts, aggregate sizing by Multisizer 4e (Beckman Coulter, Brea, CA, USA) and metabolite (glucose, lactate) analysis by Vi-Cell™ Metaflex Bioanalyzer (Beckman Coulter). After counting, cells from each sample were fixed for flow cytometric analysis as described.
Stirred suspension culture of PSCs
When viable cell density reached between 1.0 – 2.0 x 106 cells/mL in either the DASbox or BioFlo™ 320 (Eppendorf) platforms, cells were harvested, dissociated to a single-cell suspension and passaged to the next larger vessel (BioFlo 320 or Xcellerex™ XDR-10 single-use bioreactor (Cytiva, Marlborough, MA, USA)). This cell density range was chosen to align with our aggregate size limit of approximately 300 µm and was typically achieved by day 5 of suspension culture. A single cell passaging method, described below, was used. After dissociation, cells were counted and inoculated at a density of 2.5 x 105 cells/mL in either the BioFlo 320 or XDR-10. Cells were grown in mTeSR1, supplemented with 10 µM rho-associated protein kinase inhibitor Y27632, for the first 24 hours of suspension culture without perfusion. The reactors were maintained at 37°C, pH 7.2, dissolved oxygen (DO) 50%, stirred at the optimized dynamic agitation profile, and supplied with sterile-filtered air, O2 and CO2 through an overlay. 100% DO calibration was performed relative to atmospheric levels. Perfusion started at 24 hours post-inoculation with either fixed perfusion rate (50% medium refreshment per day) or enhanced perfusion, depending on the growth of cells. Perfusion in the BioFlo 320 and XDR-10 platforms was performed by a separate acoustic mini-BioSep system (Applikon Biotechnology, Delft, Netherlands). Daily sampling was performed as previously described.
Cell passaging
To passage cells between reactor vessels, aggregates were dissociated into a single cell suspension. The passaging process required five cell bags. An identical procedure was used to passage between the various bioreactor platforms (DASbox into BioFlo 320, and BioFlo 320 into XDR-10). For passage from DASBox to the BioFlo 320 we used five 600 mL bags (Baxter, Fisher Scientific) and for passage from the BioFlo 320 to the XDR-10 we used five 2 L bags (Sartorius Stedim North America, Toronto, Canada). Dissociation occurred inside of each respective bioreactor vessel. Cells destined for downstream processing were grown and then dissociated inside of the XDR-10 vessel.
Bioreactor controls such as agitation, heat and gas flow were turned off, and cell aggregates were collected into a cell collection bag, Bag #1 using air delivered through the sterile filtered exhaust line to slightly pressurize the reactor vessels. After the cells were collected, Bag #2 containing PBS (80 mL for the DASbox, 800 mL for the BioFlo 320) was sterile-welded to an input line and gravity-fed into the vessel to rinse and collect any remaining aggregates. This rinse was then added to the cell collection bag (Bag #1). The cell collection bag was then removed and brought into a biosafety cabinet. The cell suspension was then transferred into a 250 mL conical tube and the aggregates were centrifuged at 100 x g for 5 minutes. After centrifugation the supernatant was carefully decanted and the cells were washed with 250 mL of pre-warmed PBS and centrifuged again. During this time Bag #3 containing TrypLE (50 mL for DASbox, 500 mL for BioFlo 320) + 1% DNase I (v/v) (Millipore Merck KGaA Darmstadt, Germany), was infused into the reactor via sterile weld. Temperature control (37oC) was restarted and agitation was set to 80 RPM. Next, the supernatant from the centrifuged cells was removed, cells were resuspended in 50mL of PBS + 1% DNase, and loaded back into Bag #2, and infused back into the reactor. Bag #2 was left attached to the reactor vessel during this time. The reactor was operated for 10 minutes at 80 RPM to begin dissociating the aggregates. After the initial dissolution the reactor agitation was set to 100 RPM for 1 minute to completely dissociate any remaining clumps. The single-cell suspension was then collected back into the attached Bag #2 using positive pressure from air through the sterile-filtered exhaust line. Next, Bag #4 containing DMEM (150 mL for DASbox, 1L for BioFlo 320) + 1% DNase was infused into the reactor and allowed to rinse the vessel for 1 minute. The cell suspension in Bag #2 was then infused back into the reactor vessel and the contents allowed to mix to quench the dissociation reaction. The quenched cell suspension was then collected back into Bag #2, brought into the biosafety cabinet, and transferred into a fresh 250 mL conical tube. The single cell suspension was then centrifuged at 300 x g for 5 minutes. During centrifugation Bag #5 of mTeSR1 (400 mL for BioFlo 320, 2 L for XDR-10) with 10 µM rho‐associated protein kinase inhibitor Y27632 was welded on to the recipient reactor vessel, and the media was infused to begin warming with agitation at 75 RPM. The centrifuged cells were resuspended mTeSR1 media (40 mL for BioFlo 320, 300 mL for XDR-10). Next, the cell suspension was passed through a 70-micron strainer to remove any residual clumps. A cell count was performed as described. Using the procedures outlined here, cells were sequentially passaged from the DASbox into a BioFlo 320 vessel and then from the BioFlo 320 into an XDR-10 bag (thus forming a complete seed train). An inoculation density of 2.5 x 105 cells/mL was targeted across all vessels. For the first 24 hours, mTeSR1 supplemented with 10 µM rho‐associated protein kinase inhibitor Y27632 comprised the media system. After 24 hours, cells were expanded in suspension using mTeSR1 only.
Downstream processing
To concentrate and wash dissociated single cells, we harvested an XDR-10 using a closed, single-use, continuous centrifugation instrument, (Sefia™ Cell Processing System, Cytiva). First, aggregates were digested into a single-cell suspension within the XDR-10 as described for cell passaging. PBS was used throughout the downstream process to wash the cells. As a proof of concept, two separate runs with single cell suspension aliquots between 2–3 L (targeting 7 x 109 cells total) were processed using the FlexCell protocol and CT-800.1 consumable kit, washed and volume reduced to approximately 250 mL. Initially, approximately 120 mL of cell suspension was loaded into the chamber at 100 mL/min. This was reduced to 60 mL at 300 x g for 2–3 minutes. The chamber was then filled to 200 mL, spinning at 300 x g at a feed rate of 100 mL/min and the culture was concentrated for a period of 45 minutes. Once the entire input volume was concentrated, the chamber volume was reduced to 50 mL. A wash of 150 mL of PBS was added and the cells were spun at 300 x g for 10 minutes. This PBS wash and spin was repeated one additional time. Once cell washing was complete, the concentrated cells were harvested, and an additional 50 mL of PBS was used to wash the chamber for a total final volume of 250 mL.
Metabolite analysis
Cell aggregate samples were collected from culture vessels at the same time interval (every 24 hours) without interrupting the cultures and perfusion. Samples were centrifuged at 100 x g for 3 min at room temperature to remove single cells and debris. The glucose and lactate concentrations of the supernatant were analyzed with the Vi-CELL MetaFLEX™ Bioanalyzer.
Mean aggregate diameter sizing
Aggregate sizing was performed on a Multisizer 4e particle analyzer. Samples of aggregates were collected daily. Sizing was performed using a 560 mm aperture on days 1–3 of bioreactor culture, and then on a 1000 mm aperture on days 4–7 of bioreactor culture, utilizing 3 mm Hg of vacuum pressure for the 560 mm aperture, and 6 mm Hg of vacuum pressure for the 1000 mm aperture. Briefly, 2 mL of sample was added to 200 mL of Isoton II solution (Beckman Coulter) in the sample chalice. Sizing data were collected over a 40 second run time. Sizing data were exported from the supplied software as the mean of all measured events +/- standard deviation. A total of 200–1000 events were captured. A time course of mean aggregate size versus time was generated and a linear fit was applied to determine the rate of mean aggregate size increase in mm/day.
High-density seed bank preparation and application
To shorten the length of time required to carry out the 10 L PSC seed train, high-density seed banks (HDSBs) were prepared that could be used to directly seed a stirred tank reactor at the 700 mL – 1 L scale. To accomplish this, we carried out our seed train process beginning in static culture and moving through the 100 mL to 1 L and ending up at the 10 L scale. The vessel agitation was turned off, the aggregates were allowed to settle, and media was aspirated out of the vessel via a dip tube. The aggregates were then dissociated to single cells as previously described. Dissociated single cells were removed from the bioreactor into a cell bag using positive pressure from air through the sterile-filtered exhaust line, then transferred to 500 mL conical bottles for centrifugation at 300 x g for 5 minutes. The supernatant was removed, and the cells were resuspended in 7 mL of CryoStor™ CS-10 (STEMCELL Technologies) cryoprotectant at 25 x106 cells/mL, which was sufficient for direct seeding of a 700 mL volume at 2.5 x 105 cells/mL. Individual 10 mL cryovials (Fisher Scientific) were manually filled and the vials were frozen at -80 oC overnight in a CoolCell SV10 (VWR, Toronto, Canada) to ensure controlled-rate freezing. Frozen vials were subsequently placed in vapor-phase liquid nitrogen for long-term storage. Cell pluripotency was verified by flow cytometry as described. A validation expansion run was performed after the banking was completed, whereby a vial was used to directly seed a BioFlo 320 at a 700 mL volume. The target initial cell density of 2.5 x 105 cells/mL was achieved, with growth and aggregate size kinetics as expected. Cell pluripotency was verified at the end of the run by flow cytometry and a sample was prepared for karyotype analysis as described.
For the application runs in Figure 6C, a frozen vial of the HDSB was thawed and inoculated at a cell density of approximately 2.5 x 105 cells/mL into a 1 L Minibio bioreactor (Applikon Biotechnology, Delft, Netherlands) with a 750 mL working volume. Cells were expanded as described previously, although maintained at 90% DO with 100 RPM. On the third day of the expansion, the stirrer speed was increased to 120 RPM until the end of the culture. Perfusion was performed with a mini-Biosep acoustic cell retention system (Applikon Biotechnology) and was initiated 24 hours post-inoculation with a continuous fixed perfusion rate of 50% media exchange per day, as this work was performed in parallel to determination of the dynamic perfusion strategy.
Flow cytometry
PSC pluripotency was evaluated by flow cytometry. All wash steps were performed using wash buffer (2% FBS, (Thermo Fisher Scientific) and 2 mM EDTA (Life Technologies, Carlsbad, CA) in Hank’s Balanced Salt Solution (HBSS, Life Technologies)). For PSC pluripotency, aggregates were collected, digested into a single-cell suspension with TrypLE, and fixed in 2% paraformaldehyde (PFA, Electron Microscopy Sciences, Hatfield, PA) for 10 min at room temperature. Samples were then washed in wash buffer (filter sterilized 2% FBS-HBSS with 2 mM ETDA) and stored at 4°C until stained. For the evaluation of cell-surface and intracellular pluripotency markers, 2 x 105 cells were prepared in V-bottom plates and permeabilized with a 0.1% (v/v) solution of wash buffer with Triton™-X 100 (Sigma Aldrich, St. Louis, MO, USA) by incubating for 5 min at room temperature. For evaluating PSC pluripotency, cells were next stained with antibodies against Nanog, Oct3/4, Sox2, SSEA-4 (BD Biosciences, San Jose, CA, USA), and Tra-160 (Biolegend, San Diego, CA, USA) diluted in permeabilization buffer (filter sterilized 2% FBS-HBSS with 2 mM ETDA and 0.1% v/v Triton X-100).
Single-stained compensation beads (BD Biosciences) were prepared in parallel. Compensation controls and stained samples were incubated for 30 min at room temperature, in the dark. Unstained cell samples were incubated with permeabilization buffer alone. After staining, sample plates were centrifuged at 500 x g to pellet the cells, and the supernatant was removed. Samples were washed twice in wash buffer and then analyzed on a CytoFlex™ flow cytometer (Beckman Coulter). Flow cytometry data were analyzed by FlowJo™ software (BD Biosciences).
Karyotyping analysis
Cell lines were routinely subjected to karyotype analysis at the end of the suspension expansion protocols. Cells were prepared according to the requirements of the Cambridge University Hospital Cytogenetics Laboratory (Cambridge, UK). Briefly, at the end of the seed train, cell aggregates were dissociated as previously described and plated onto Matrigel-coated 6-well tissue culture plates. Cells were cultured as previously described with daily media exchange (see maintenance of PSC in 2D culture) until 50 – 70% confluent. At this point, KaryoMAX™ Colcemid in PBS (Thermo Fisher Scientific) was added to the growth media to make a final concentration of 100 ng/mL, and the plate was incubated for 1 hour under normal culture conditions. After incubation, the culture supernatant was collected, the cell monolayer was washed with PBS, and the wash was collected with the supernatant. Cells were then detached from the wells using 1 mL TrypLE Select per well and incubation for 4 minutes at 37°C. Cells were triturated into a single cell suspension, and the dissociation reaction was quenched with mTeSR1 media. After centrifuging at 200 x g for 5 minutes and aspirating off the supernatant, the cell pellet was resuspended in 300 µL of fresh media. Next, 10 mL of 0.075 M KCl solution (KaryoMAX KCl solution, Thermo Fisher Scientific) was added dropwise to the cell suspension while vortexing. The cells were then incubated at room temperature for 20 min to allow swelling. After this incubation, cells were centrifuged at 200 x g for 5 minutes, and the KCl solution was aspirated. A 3:1 solution of methanol (Thermo Fisher Scientific) and glacial acetic acid (Thermo Fisher Scientific) was made in-house and used to fix the cells. The fixative was added dropwise to the test tube with simultaneous vortexing. The cells were centrifuged again, and the fixative removed. The cells were then resuspended in 2 mL of fresh fixative solution and stored at -20oC until analyzed. Samples were sent to Addenbrooke’s Hospital, Cambridge University Hospital Cytogenetics Laboratory, for analysis.
Results & Discussion
Parameter optimization for a 10 L seed train
We set out to develop a cell expansion protocol, or seed train, to generate > 1010 human pluripotent stem cells (hPSC) in the Xcellerex 10 L single-use stirred bioreactor (XDR-10), as this platform has a scalable, incremental path to 2000 L and a history of GMP manufacturing. We performed a survey of small-scale (<10 L), single-use platforms suitable for GMP manufacturing [20]Pigeau G, Razvi A, Huang S. Predicting bioreactor product production based on independent or multivariate analysis of multiple physical attributes. 2018; US 62/665,155.Pigeau G, Razvi A, Huang S. Predicting bioreactor product production based on independent or multivariate analysis of multiple physical attributes. 2018; US 62/665,155., [21]Pigeau GM, Csaszar E, Dulgar-Tulloch A. (2018). Commercial Scale Manufacturing of Allogeneic Cell Therapy. Front. Med. 2018; 5: 233.Pigeau GM, Csaszar E, Dulgar-Tulloch A. (2018). Commercial Scale Manufacturing of Allogeneic Cell Therapy. Front. Med. 2018; 5: 233. and selected the Eppendorf DASbox (60 - 250 mL) and BioFlo 320 (400 – 1000 mL) as the intermediate steps to the 10 L scale, with the DASbox acting as the small volume, de-risking platform.
The seed train was developed starting with the DASbox bioreactor at 160 mL, passaging to the BioFlo 320 platform at the 1 L scale, which generated enough cells to seed the XDR-10 at an 8 L operational volume. To ensure successful expansion at each scale, set points for fundamental process parameters for PSC suspension culture were determined, using studies performed in small volume DASbox bioreactors and with a commercially available hESC line (ESI-017) as a model. In these initial experiments, agitation, DO, and perfusion feeding strategy were investigated, and enabling profiles for each parameter were specified.
With single-use, GMP use as the driver for platform selection, we encountered differences in vessel geometry/aspect ratio, volume turndown ratios and impeller design. Traditional bioprocess scale-up parameters such as mixing time, power input per volume (P/V), oxygen mass transfer (kLa), tip speed (Vtip), impeller to reactor diameter ratio (D/Tv) were calculated and reviewed to guide the process development. However, these were not always directly applicable due to the platform differences and the unique constraints and characteristics of culturing PSCs in stirred tanks (for example, aggregate size limitations, shear sensitivity, quality concerns, downstream differentiation ability). Therefore, rational experimental designs to explore the operational space were utilized as required in the small volume de-risking platform.
Agitation rate was first investigated in the DASbox bioreactors. Initial experiments were performed to determine the minimum enabling agitation rate, sufficient to maintain a suspension culture. This was achieved by testing increasing agitation rates of 30, 45, 65 and 80 RPM. It was demonstrated that a rate greater than 65 RPM was required to maintain the PSC aggregates in suspension culture. Agitation rates of less than 65 RPM resulted in aggregate settling (data not shown). A second study with agitation setpoints of 75, 80, and 85 RPM as well as with a dynamic strategy of increasing agitation at 75, 80, and 85 RPM on days 0, 1, and 2, respectively, was performed to determine if a fixed or dynamic stirring strategy would be more suitable. In this experiment, cells were cultured in the DASbox bioreactor system at 160 mL, with one vessel for each of the four agitation conditions, for five days per passage. Data from the first passage were not included, as the cells were adapted to suspension culture. Data from passages 2–5 were collected and are depicted in Figure 1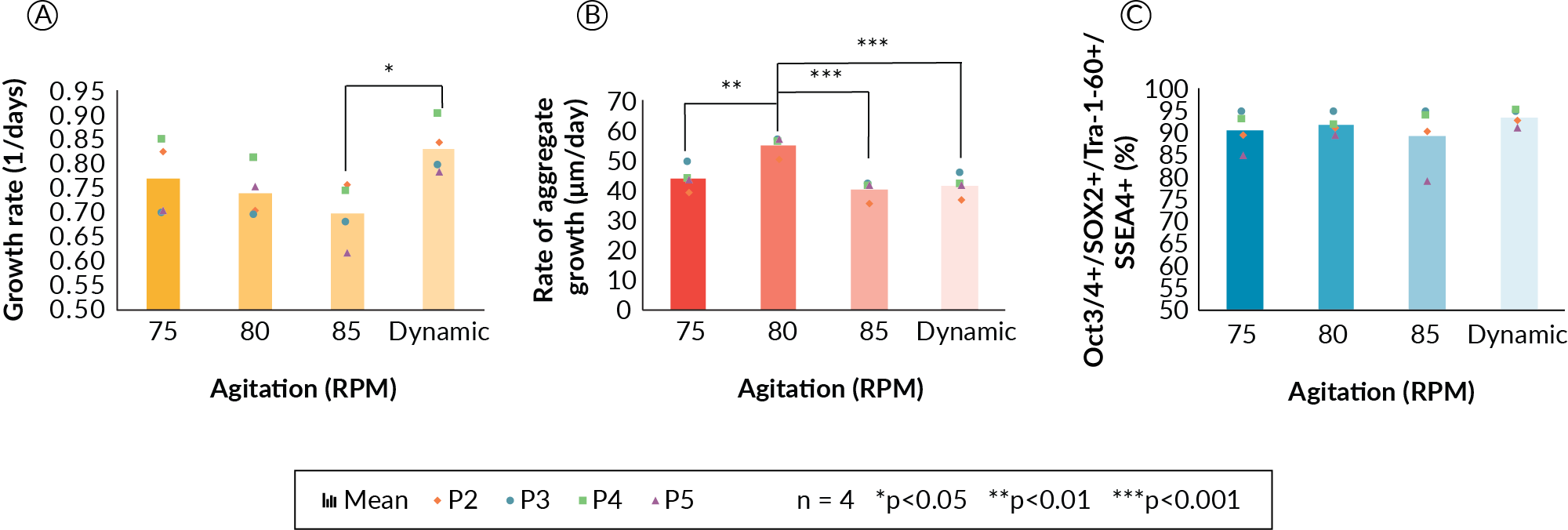
It was hypothesized that a dynamic mixing strategy would be most beneficial for PSC suspension culture because a lower agitation rate at the beginning of the culture would promote aggregation and enhance cell survival during the initial stages of the culture, and an increase would be beneficial to maintain suspension as cell aggregates grew in size. We also sought to use agitation to control aggregate size as the culture expanded. In order to maintain adequate and biologically relevant oxygen availability to PSCs in aggregates, we referred to suggested oxygen diffusion limits from blood vessels of approximately 100–200 µm [5]Chen KG, Mallon BS, McKay RDG, Robey PG Human pluripotent stem cell culture: considerations for maintenance, expansion, and therapeutics. Cell Stem Cell 2014; 14, 13–26.Chen KG, Mallon BS, McKay RDG, Robey PG Human pluripotent stem cell culture: considerations for maintenance, expansion, and therapeutics. Cell Stem Cell 2014; 14, 13–26.. With this in mind, we aimed to develop an agitation scheme able to control the mean aggregate diameters to below 300 µm on the day of cell harvest. These experiments demonstrated that PSC cellular growth rate (Figure 1A) may be affected along with the rate of aggregate size increase at 85 RPM and in the dynamic agitation profile (Figure 1B).
Expansion kinetics (growth rate, Figure 1A) decreased with increasing agitation rate, but did not show statistical significance across the fixed agitation conditions in our experiments. However, the growth rate was significantly lower for the 85 RPM condition as compared to the dynamic condition (p < 0.05). Presumably, 85 RPM imparted a higher shear during the initial aggregation, which may have prevented formation of aggregates and resulted in slower growth. In general, a trend of decreasing growth rate was observed as the agitation rate was increased. The rate of aggregate growth was significantly higher for the 80 RPM condition (Figure 1B) as compared to all other conditions, but interestingly, the rate of aggregate size increase does not correlate with specific growth rate. The 80 RPM condition stands alone and in contrast to the three other conditions in this respect. More experiments would be required to elucidate the nature of the rate of aggregate size change at the 80 RPM condition. Despite differences in growth kinetics and aggregate size, cells displayed high levels of viability and pluripotency maker expression for all four mixing conditions (Figure 1C). Greater than 91% of the cell population was four-marker positive (Oct3/4, Sox2, Tra- 1–60 and SSEA-4—as assessed by flow cytometry) in all cases, and the dynamic agitation strategy produced the highest pluripotency data at 95% four-marker positive. From the experiments completed, the dynamic mixing strategy was carried forward as it gave the highest growth rate, did not negatively impact aggregate size increase, and cells retained a high level of pluripotency marker expression.
The second parameter investigated was DO, which are related to the percent of oxygen saturation at 37 °C and normal pressure. DO has physiological [5]Chen KG, Mallon BS, McKay RDG, Robey PG Human pluripotent stem cell culture: considerations for maintenance, expansion, and therapeutics. Cell Stem Cell 2014; 14, 13–26.Chen KG, Mallon BS, McKay RDG, Robey PG Human pluripotent stem cell culture: considerations for maintenance, expansion, and therapeutics. Cell Stem Cell 2014; 14, 13–26., diffusional and cost implications in large-scale suspension culture. Thus, understanding the impact of DO and identifying the minimum DO necessary to maintain growth rate and pluripotency will impact scaling strategy and may impact manufacturing costs at scale. This is especially important for cell types that are particularly shear sensitive and consequently require obligate headspace gassing as opposed to sparging, such as PSCs [16]Nienow AW, Coopman K, Heathman TRJ, Rafiq QA, Hewitt CJ (2016). Bioreactor Engineering Fundamentals for Stem Cell Manufacturing. In Stem Cell Manufacturing, J.M.S. Cabral, C.L. da Silva, L.G. Chase, and M.M. Diogo, eds. (Elsevier), 2016; 340..
In this experiment, a single DASbox was cultured at 90% DO (passage 1). This DASbox generated enough cells to split into four DASboxes at DO settings of 10, 30, 50, and 90%, respectively. Each DASbox was run for three further passages. The first passage at each DO condition (passage 2) was taken as an adaptive one, and the final two passages were used for comparative data analysis (passages 3 and 4). The cells from the DO study were then pooled and used to seed a 1 L BioFlo 320 vessel, which was then passaged to the first XDR-10 run (Seed Train 1).
Viable cell density (VCD) data from passages 3 and 4 (Figure 2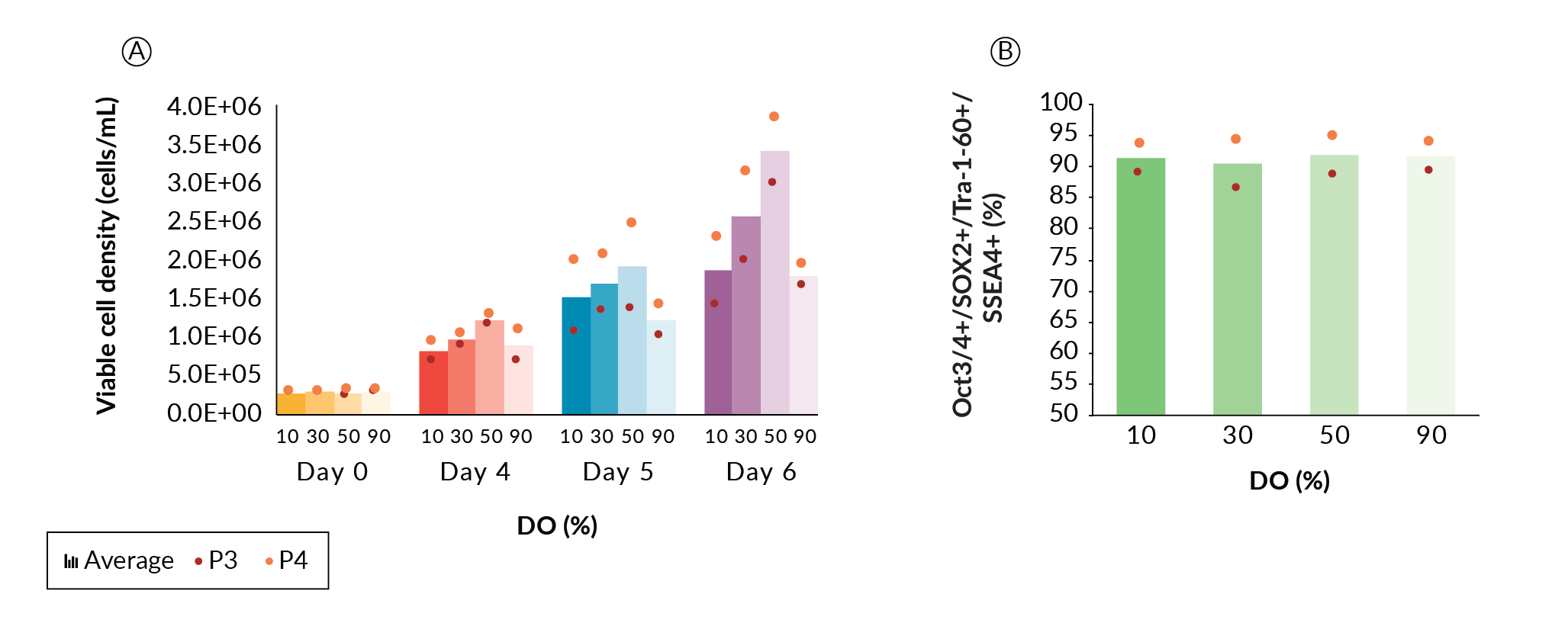
The final parameter optimized was perfusion feeding strategy. Perfusion was chosen as the culture mode to continuously supply temperature-labile media components such as growth factors and cytokines, and to wash out inhibitory byproducts such as lactic acid. We examined the differences and effects of a fixed versus dynamic/responsive perfusion strategy on PSC growth and quality. Four DASbox reactors, two with fixed perfusion rate and two with dynamic perfusion, were run. Perfusion was initiated at 50% of the culture volume per day on day 1, on the condition that aggregation was confirmed microscopically for all bioreactors. From this point, whenever referring to the perfusion rate, it is a percentage of the culture volume per day unless otherwise stated. The two reactors with fixed perfusion conditions remained at 50% for the remainder of the culture. For the two dynamically perfused bioreactors, we chose to increase perfusion rate when pH decreased to 6.8 (typically day 3 or 4). When this pH drop was reached, perfusion rate was manually increased by 30% over the previous set point. From that point forward, 30% increases from the previous set point were made daily (50%, 80%, 110%...etc). We did not optimize the point or rate of perfusion increase further but suggest there would be additional operational space to examine. Figure 3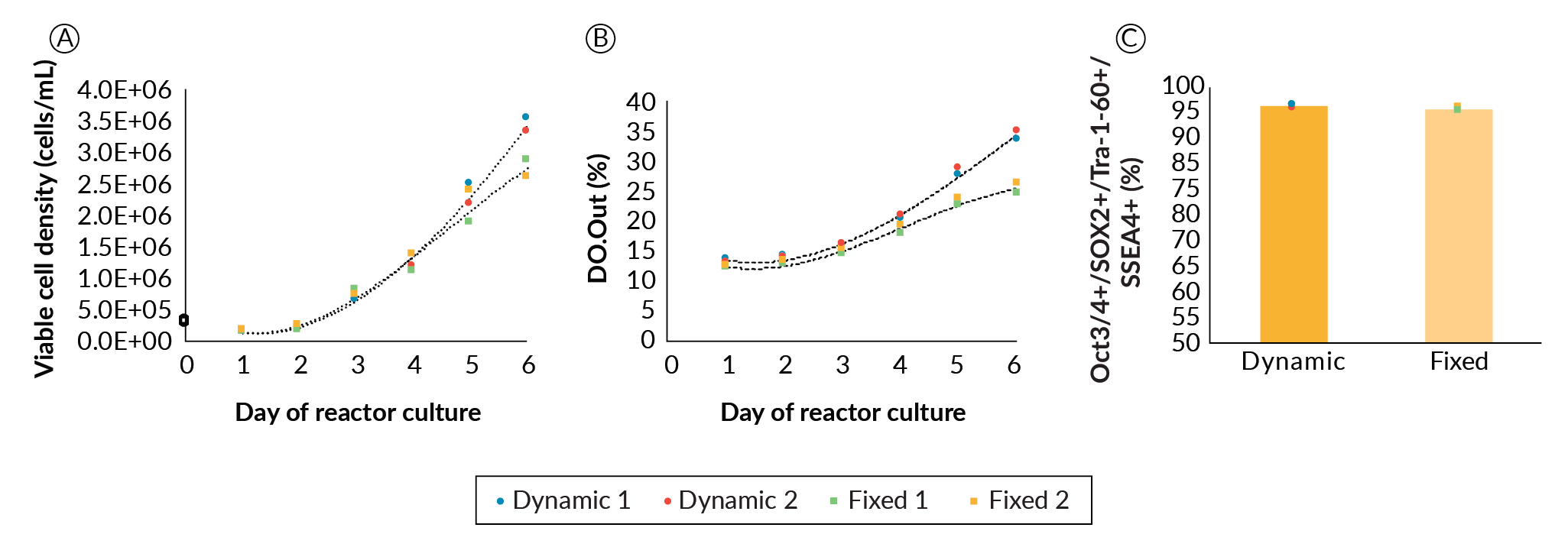
In the fixed perfusion conditions, lactate concentration reached 15 mM on day 4 and continued to climb to 16 mM by day 6. Dynamic perfusion kept lactate concentration below 15 mM (data not shown). In parallel, glucose level dropped to 4.6 mM and then 2.7 mM, respectively in these reactors (data not shown). As high levels of inhibitors and low levels of nutrients generally do not favor PSC growth [11]Kropp C, Kempf H, Halloin C, et al. Impact of Feeding Strategies on the Scalable Expansion of Human Pluripotent Stem Cells in Single-Use Stirred Tank Bioreactors. Stem Cells Transl. Med. 2016; 5: 1289–1301.Kropp C, Kempf H, Halloin C, et al. Impact of Feeding Strategies on the Scalable Expansion of Human Pluripotent Stem Cells in Single-Use Stirred Tank Bioreactors. Stem Cells Transl. Med. 2016; 5: 1289–1301.Kropp C, Kempf H, Halloin C, et al. Impact of Feeding Strategies on the Scalable Expansion of Human Pluripotent Stem Cells in Single-Use Stirred Tank Bioreactors. Stem Cells Transl. Med. 2016; 5: 1289–1301., it was hypothesized that growth rate would improve if the perfusion rate was increased throughout the expansion. While levels of glucose and lactate may affect expansion, there is insufficient data to determine whether these alone are responsible, due to provision of other growth factors, cytokines and nutrients and dilution of other potentially inhibitory by products as the perfusion rate is increased. Nonetheless, the higher rate of media exchange in the dynamic perfusion strategy promoted better cell expansion comparatively, which became noticeable from day 4. On day 6, dynamic perfusion led to an average 25% increase in cell density compared to the fixed perfusion rate bioreactors. Despite ample potential for further optimization, we progressed with the current perfusion strategy.
The influence of agitation, DO, and perfusion rate on the expansion of ESI-017 as aggregates in process-controlled bioreactors was studied. Based on these data, a protocol for the expansion was developed. A set point of 50% for DO, a dynamic agitation profile starting at 75 RPM upon inoculation and increasing by 5 RPM/day to 85 RPM on day 2, and a perfusion strategy starting at 50%/day on day 1 with rate increases of 30% over the set point every day once culture pH first drops to 6.8 was specified. These parameters were carried forward and formed the basis for further scale-up work.
Application of the optimized parameters to the expansion of a hiPSC line
Once the agitation, DO and perfusion strategies and setpoints were specified, the robustness of the process was evaluated using a second PSC line. The human induced pluripotent stem cell (hiPSC) line NCRM1 was compared to the human embryonic stem cell (hESC) line ESI-017 used previously. Both ESI-017 and NCRM1 were suspension-cultured in one DASbox each for a single passage. Each cell line was then split into two bioreactors. The cells were cultured in DASbox bioreactors for three additional passages. From this, a total of seven passage datasets were collected. Datasets contained growth, viability, metabolite, and aggregate size kinetic data as well as pluripotency.
While we agree that passage-based data is not a sufficient replacement for biological replicates, this approach demonstrates the stability of the process across the multiple passages required to scale the expansion process. From these data, the two cell lines showed no significant differences in growth rate, and no metabolic differences were noted in terms of lactate yield from glucose (Figure 4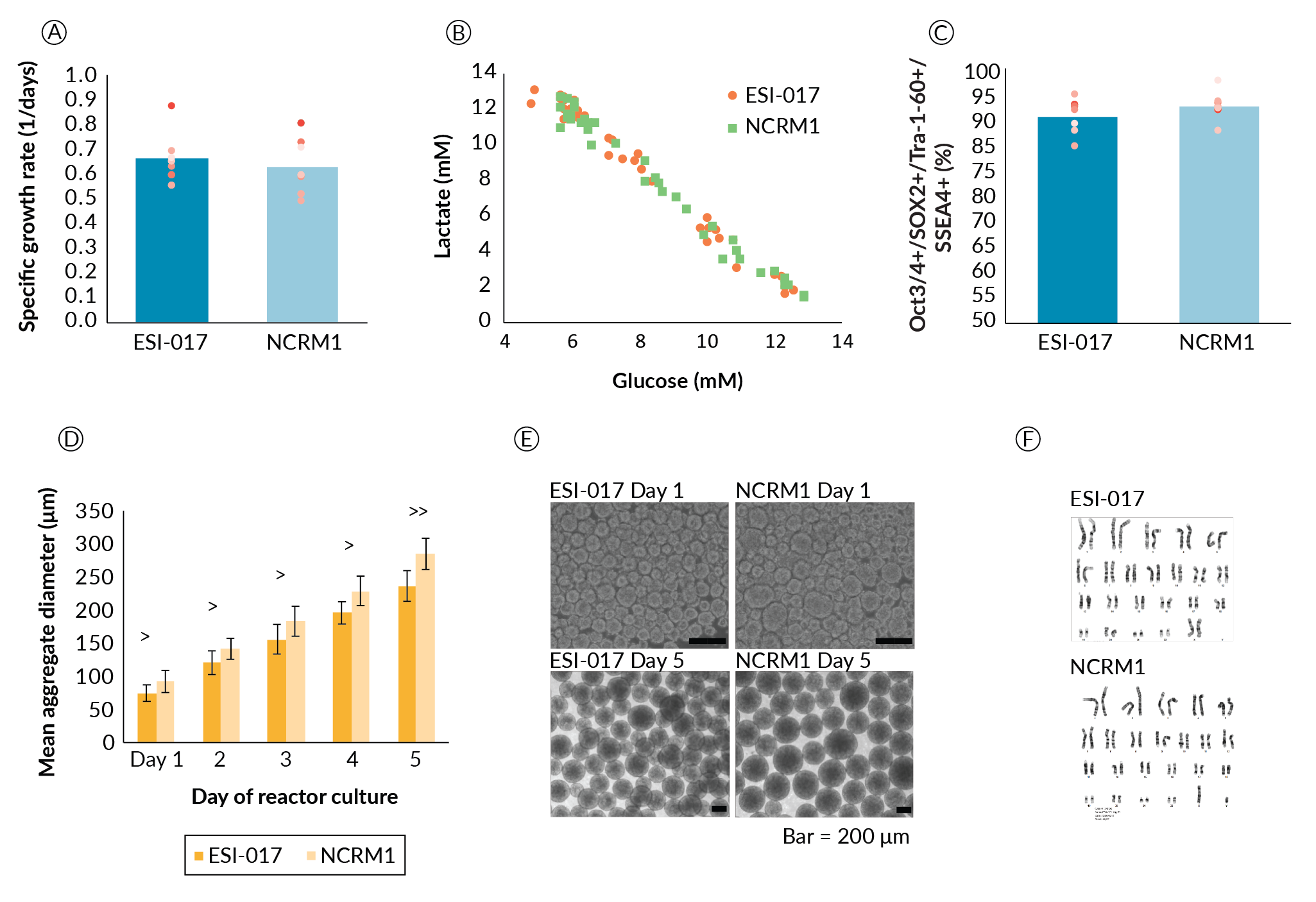
Aggregate formation appeared to be different between the two cell lines (Figure 4D and Figure 4E). Aggregate size was significantly different on day 1 with ESI-017 cells forming aggregates with an average diameter of 74.9 µm and NCRM1 cells forming aggregates with an average diameter of 92.4 µm (p < 0.05). This size difference persisted throughout the culture duration. When we quantified the rate of aggregate size growth, this difference narrowly missed achieving significance at the α = 0.05 level (p = 0.062). ESI-017 aggregates grew at an average rate of 41 µm/day compared to NCRM1 at 48 µm/day. It is hypothesized that the difference in the initial aggregate formation could be due to cell line differences in response to the shear experienced on transition to suspension culture.
Despite the small differences in the rate of aggregate size increase, we proceeded with the ESI-017- determined process parameters for scale-up of NCRM1. For cell lines that are not successful following this scale-up protocol, we propose that day 1 aggregate sizing may prove to be a key datapoint highlighting differences that could influence process parameters such as agitation and culture length due to the potential impact of aggregate diameter on aggregate settling and oxygen availability. With respect to process robustness, aggregate formation and day 1 aggregate size may prove important sources of line-to-line variability and may be key parameters when transferring additional cell lines to this process.
Correlation of hPSC growth kinetics with stirred tank bioreactor
process parameters
As PSCs expand, they produce lactate and consume oxygen and glucose. Correlations between process values and outputs from installed pH and DO probes were investigated with discrete sample data such as viable cell density, lactate production and glucose consumption. Any lactate and CO2 produced will acidify the medium, thereby lowering the pH. As DO was being controlled, the rate of change of the probe output signal (DO.Out), which reflects the culture’s demand for oxygen, was also investigated for correlation to growth.
Growth data from the ESI-017 and NCRM1 comparison study were plotted against both pH and DO probe output (Figure 5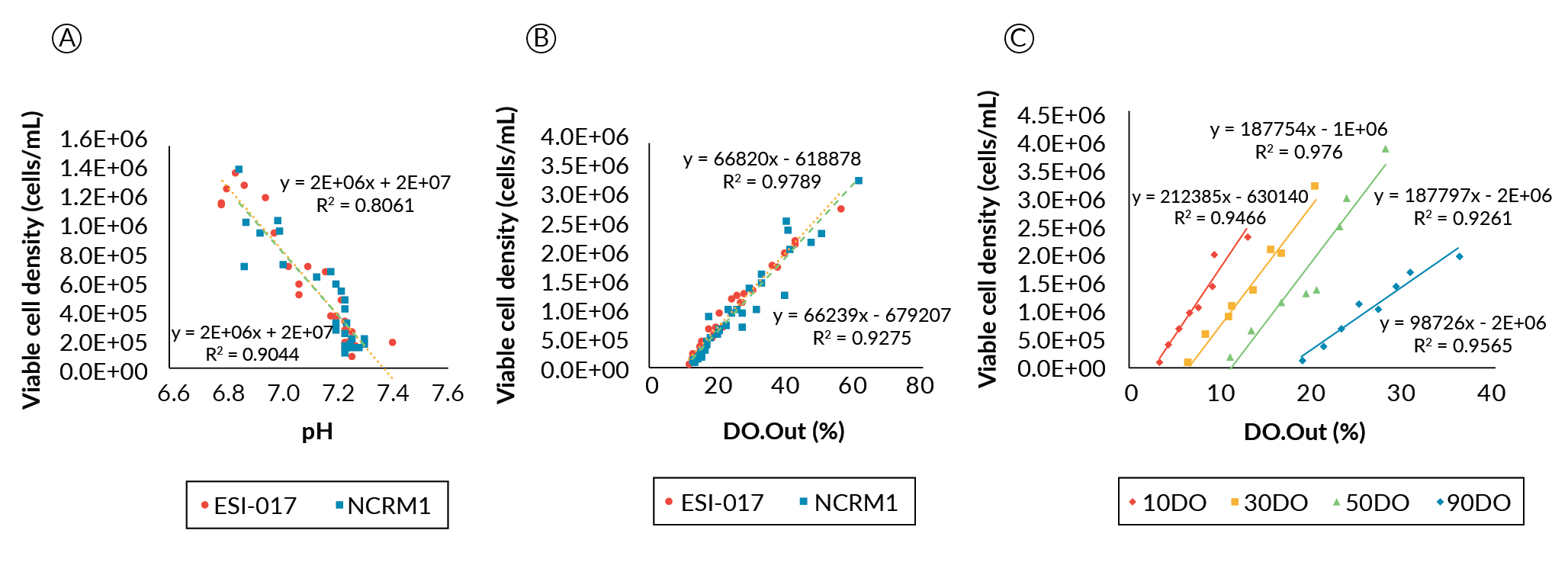
We additionally investigated if a good linear correlation could be found for the cells cultured at different DO setpoints from those used in the preliminary DO determination study (Figure 5C). Among the different DO conditions, DO90 had a shallower slope compared to the low DO conditions, whose slopes appeared essentially parallel. We do not believe this to be a physiological response, but rather it may be related to a reduction in driving force to dissolve oxygen as the saturation point is approached at the higher setpoint; whereas the parallel linear correlations at the lower DO set points indicate that the oxygen demand to cell density correlation in this system is unaffected at ≤ 50% DO.
Taken together, the correlation analyses suggest that both pH and the oxygen demand value from the online probes can be used to estimate the hPSC growth condition in the bioreactors. Though the pH correlation can be linked to the viable culture density irrespective of the bioreactor geometry, it will be impacted by how fast medium is exchanged and the buffering capacity of the medium. On the other hand, DO correlation is less influenced by the perfusion rate and the medium type, but will be influenced by the parameters that will affect the oxygen transfer coefficient. Interestingly, different DO conditions are likely to impact the oxygen consumption rate of the cells. Therefore, to have a more accurate estimation of viable cell density in the bioreactor, both pH and DO probe signals should be considered and analyzed.
Since we have established a correlation between the bioreactor process parameters and viable cell counts, we propose the dynamic feeding regime could be cascade-controlled via integration with the pH and DO continuous process data [20]Pigeau G, Razvi A, Huang S. Predicting bioreactor product production based on independent or multivariate analysis of multiple physical attributes. 2018; US 62/665,155.Pigeau G, Razvi A, Huang S. Predicting bioreactor product production based on independent or multivariate analysis of multiple physical attributes. 2018; US 62/665,155., [21]Pigeau GM, Csaszar E, Dulgar-Tulloch A. (2018). Commercial Scale Manufacturing of Allogeneic Cell Therapy. Front. Med. 2018; 5: 233.Pigeau GM, Csaszar E, Dulgar-Tulloch A. (2018). Commercial Scale Manufacturing of Allogeneic Cell Therapy. Front. Med. 2018; 5: 233.. This would enable metabolic feedback of the system to control feed rates, providing a level of physiological control and automation of the manufacturing process. Additionally, with proxy measures of viable cell density via pH and DO correlations, the need to extract samples from the bioreactor during manufacturing would be removed, reducing labor and contamination risk.
Scale-up of suspension aggregate culture to 1 L and 10 L reactors
Parallel to the DASbox process development efforts, scale-up was also being performed at the 1 L BioFlo 320 scale and the 10 L XDR-10 scale. As process improvements were being realized in the DASbox bioreactors, they were being applied to the scale-up efforts occurring in parallel. Numerous parameters were evaluated in the scale-up activities including agitation, DO and pH control strategies, perfusion strategy, closing of the passaging and harvest process, and demonstration of scalability. We found that agitation, DO and perfusion rates scaled nearly directly between the DASbox and BioFlo 320 platforms, with only a small decrease in agitation required to maintain mixing time in the 1 L BioFlo 320. Agitation rates to maintain mixing time in the XDR-10 were reduced even more, due to more significant differences in platform and impeller design. A total of 9 expansion runs at the 10 L scale were made. Two critical quality attributes (CQA) for passaging have been identified, with aggregate size being primary and cell number being secondary.
The first attempt at the 10 L scale was seeded from the cells generated as part of the DASbox DO study. Cells from all four DASboxes were pooled and used to inoculate a single BioFlo 320. The BioFlo 320 was cultured for 7 days generating enough cells to seed an XDR-10 at 5.5 L at the target seeding density of 2.5 x 105 cells/mL. A perfusion technology solution had not been developed at this point, so a combined fed-batch/perfusion-based approach was used to enable the culture. Despite not having an ideal culture mode, a total of 7.75 billion cells were generated. Perfusion was required to maintain low levels of inhibitory by-products, but the Applikon BioSep used could only achieve 1500 mL/day, which is 28% volume/day at the starting volume and 19% volume/day at the final volume. It was demonstrated earlier that 50% volume/day increases in perfusion rates were required to maintain exponential growth. To overcome this, mTeSR1 supplement was bolus-fed into the XDR-10 on days 2, 3 and 5 using 500 mL for the first two feeds and 1750 mL for the last feed, resulting in a total volume of 8250 mL.
Seed trains 2, 3 and 4 were executed to identify a perfusion solution for the XDR-10. Seed train 2 used the dip tube in the XDR-10 Pro Plus bag as a gravity settling device. The intent was to allow the heavier aggregates to be retained while spent media was removed. This was not successful as significant aggregate loss was observed in the waste media collection. Seed train 3 used the Applikon 10 L BioSep, which has a larger capacity, which is not an in-bag solution but rather a retrofit. It is also more complicated and requires two pumps to operate. The first is a higher flow-rate pump to supply the BioSep with culture and the second removes the waste. Despite multiple attempts to configure the setup correctly, aggregates settled at various low points in the setup. Additionally, continuously exposing PSC aggregates to a peristaltic pump head is not advisable, as significant viability loss and growth impacts were observed (data not shown). Seed train 4 utilized Repligen’s ATF (alternating tangential flow filtration) system. Based on in-house, single-pass shear assessments of aggregates through hollow fibers in a tangential flow filtration (TFF) system, and recommendations made by the vendor, a shear rate of 2700 1/s was used to recirculate the culture and remove waste via perfusion. This perfusion set-up was abandoned due to adverse impact on cell viability and, like the high flow rates in seed train 2, was hypothesized to be too harsh for PSC aggregates.
To date, a commercially available, large-scale, single-use solution for perfusion of PSC aggregates has yet to be developed. As such, a prototype perfusion device for PSC expansion was developed and tested. PSCs grown as aggregates in bioreactors have a reasonably high settling velocity, but not quite enough to resist removal through a dip tube. Based on this, a perfusion device composed of spiral tubing coupled with a top-mounted 20 µm filter was devised. Through inclusion of a filter element and a pump having a flush-back cycle, the aggregates are preferentially returned to the bioreactor environment while waste media is removed. To demonstrate the device’s utility, NCRM1 cells, grown as part of an iPSC experiment to demonstrate robustness of the expansion protocol, were passaged to a 1 L BioFlo 320 retrofitted with this settling device, twice (P2 and P3). Cells were then passaged a fourth time (P4) in the BioFlo 320 and in parallel, to an XDR-10 also retrofitted with the same settling device.
The BioFlo 320 was seeded at 2.5 x 105 cells/mL at 700 mL with an agitation setting of 70 RPM on day 0; culture volume was increased to 1 L on day 1 and agitation rate increased to 75 RPM once aggregation was confirmed. Perfusion was started on day 1. On day 2 the agitation rate was increased to 80 RPM.
To determine if NCRM1 aggregate morphology was impacted by use of the settling device, it was run only intermittently during the first passage in the BioFlo 320. Samples of the culture were taken before and after use of the device and were analyzed microscopically, with no observable impact on aggregate morphology. The culture was then passaged two additional times (P2-P3) into the same BioFlo 320, now with the settling device running in continuous mode for the entire expansion. Pluripotency was examined via flow cytometry after each of P1, P2 and P3 and was found to exceed our minimal requirements (97%, 94% and 93%, respectively). On the final passage (P4), the 1 L culture was used to seed the XDR-10 bioreactor (seed train 1). Pluripotency for the P4 expansion in the XDR-10 was 82% positive for the four-marker panel. We recognize the downward trend of pluripotency seen for NCRM1 through these passages, however we have not seen this for ESI-017. Additionally, on previous, repeated passaging of NCRM1 (Figure 4C) we did not observe a linear decrease in potency through P2-P4 in bioreactor culture (replicate 1: 93%, 93%, 92% and replicate 2: 88%, 93%, 97%).
The XDR-10 working volume was 8 L. Agitation speed was 55 RPM on day 0, increased to 60 RPM on day 1 and 65 RPM on day 2 with perfusion starting on day 1. To enable the back flush of the aggregates accumulating in the settling filter, the waste pump was placed under the control of the BioSep controller. Enabling settings for the back-flush time and overall cycle frequency were identified to be 10 seconds and 7 minutes, respectively. Masterflex L/S size 25 tubing was used for the spiral settling tubing. For the BioFlo 320 a single spiral tubing setup was used while for the XDR-10 a double spiral setup was applied. Masterflex L/S size 16 tubing was used in the pump head for the BioFlo 320 expansions and Masterflex L/S size 25 tubing for the XDR-10 expansion. Figure 6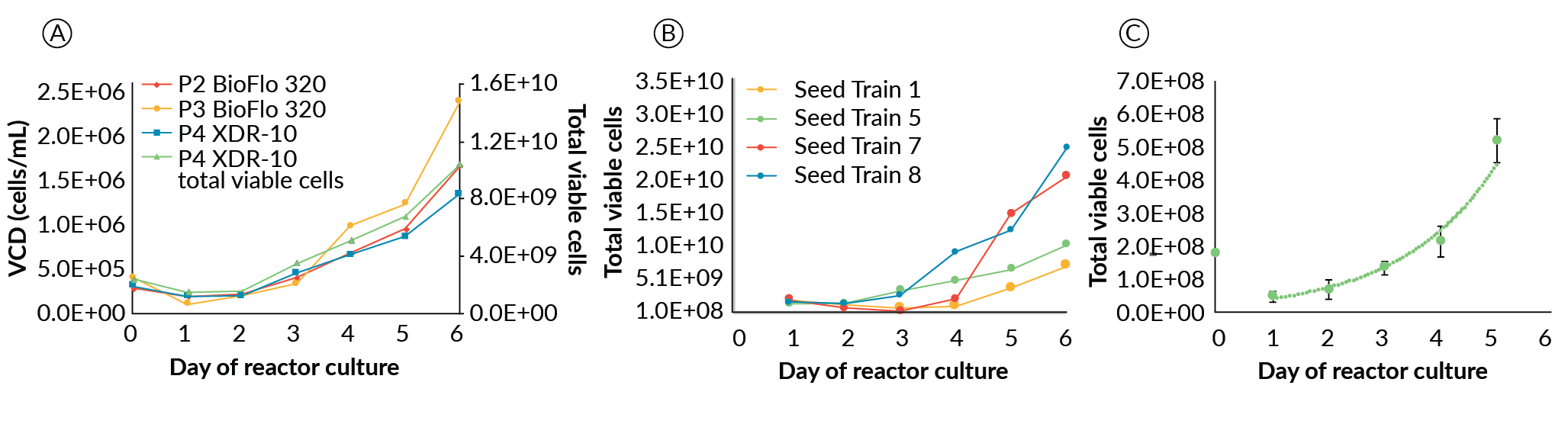
Scale-Up to the XDR-10
For the successful ESI-017 XDR-10 runs (seed trains 1, 7 and 8), differences in growth at increasing scale were observed; however, this did not prevent growing over 10 billion cells at the XDR-10 scale. Seed trains 7 and 8 exceeded our target (25 and 37 billion PSC’s, respectively); whereas the first, which did not have a suitable perfusion solution, was slightly short of our goal (7.7 billion PCSs). The NCRM1 scale-up run, from development of the large-scale perfusion device (seed train 5), also exceeded our target by producing 15 billion PSCs. The four successful scale-up runs in the XDR-10 (Figure 6B) maintained acceptable pluripotency (86%, 82%, 81% and 97% for seed trains 1, 5, 7 and 8, respectively) with no karyotypic abnormalities.
The cause of variability in final pluripotency from the 10 L runs is unknown. Certainly, these scaled up expansions describe developmental work, and the standard operating protocols for clinical manufacturing have not been locked down. However, the state of analytics (flow cytometry and karyotype analysis) and their suitability for application to cell populations in the billions deserve some consideration. Nonetheless, we considered these runs successful as they were above our internal CQA for pluripotency (≥ 80% positive for Oct3/4, Sox2, SSEA-4 and Tra-160) and returned normal chromosome G-banding results compared to master cell banks.
A drop in viable cell density is observed across all expansions regardless of scale or culture platform during the first 24–48 hours. We hypothesize this is a result of multiple stress factors as opposed to a dilution effect associated with a volume change due to initiation of perfusion or bolus feed of media, as both viable cell density and total cell number are shown to drop (Figure 6A). Harsh passaging conditions (treatment with TrypLE, trituration, multiple centrifugation steps, etc.), acclimatization to a new cell culture environment, and efficiency of aggregate formation (i.e. not all of the inoculated single cells will form aggregates), may all contribute to loss of viable cell density in the initial phase.
Perfusion was started in all cases on day 1, and rates were increased at 30%/day of the previous set point, once the culture pH dropped to our internal target of 6.9. The pH did not drop below 6.8 throughout any of the expansions. Lactate did not exceed 14 mM at any scale. The average growth rate across the two ESI-017 seed train runs with the prototype perfusion device (seed trains 7 and 8) was 0.761 + 0.131/day. Average lactate yield was 1.533 + 0.162 mol lactate produced/mol glucose consumed. The average rate of aggregate size increase was 32 + 12 mm/day.
As a final note on the last scale-up run performed, this process was accomplished completely closed, with in-reactor aggregate dissociation and tube-welded transfers. The intent was to demonstrate that large-scale PSC expansion is possible in a completely closed system and to provide a potential starting point for large-scale, clinically relevant production of pluripotent stem cells. We recognize there are many potential opportunities to improve, and to continue scaling of this manufacturing workflow, and by no means present a final commercial-ready solution. However, we feel with the scale achieved and approach taken, this work represents the most advanced biomanufacturing process for PSCs reported to date.
High-density seed bank for direct inoculation of 1 L stirred tank bioreactor
With success reaching > 1010 cells produced in the 10 L Xcellerex platform, we hypothesized these cells could be banked at high density for use as an inoculum, as in traditional bioprocesses. Cryogenically preserved, high-density seed banks (HDSB) were made to target the cell numbers required to directly inoculate a 1 L bioreactor at 2.5 x 105 cells/mL. The HDSBs had a post thaw viability of 89.1 +/- 1.9 % and were successfully used to inoculate seven 1 L bioreactors, resulting in very consistent growth rates with an average of 0.59 +/- 0.1 day-1 (Figure 6C). These growth rates were not significantly different (p = 0.569) than those in the cell line comparison study for ESI-017 of 0.66 +/- 0.1 day-1. However, both growth rates were slightly lower than the average of the two 10 L runs (0.633 day-1), likely due to differences in perfusion technologies and overall reactor set-up and operation. Day 5 pluripotency in the HDSB expansions averaged 87.6 +/- 12.6% positive for the four-marker pluripotency flow cytometry panel. While the average was above our target CQA of 80% positive, we noted one replicate experiment was unusually low at only 64% positive. The cause of low pluripotency from this run is not known and unexpected, as the bioreactor run conditions were held constant for all HDSB – 1 L expansions. On removal of this outlier, the day 5 pluripotency averaged 91.8 +/- 6.4%. Whether or not this data point was excluded, the HDSB pluripotency was not significantly (p = 0.624 or 0.578) different than that found in the cell line comparison work at 90.4 +/- 3.5%.
We did note differences in the amount of cell loss following inoculation. With the tissue culture inoculated expansion, cell counts reduced by 50% on day 1 compared to a 70% loss on day 1 in the HDSB inoculated expansions. Operationally, we note that the HDSB expansions were perfused at 50% per day and were not fed with the dynamic perfusion regime developed herein. Together, as a result, the day 5 cell counts for the cell line comparison study were on the order of 106, while the HDSB expansions only reached 105 in the same amount of time. Nevertheless, we feel that the maintenance of specific growth rate and end-point pluripotency positions the use of HDSBs as an appropriate replacement for tissue culture-based inoculum for bioreactor PSC expansion.
The decoupling of adherent tissue culture and small-scale (200 mL) stirred tank bioreactor expansion from large-scale (1 L) stirred tank bioreactor expansion is a significant advancement in the PSC manufacturing paradigm. The manual, adherent tissue culture process requires a high level of skill and consistent technique. With the use of HDSB as inocula, this source of process variability can be eliminated through normalization of cellular input. Additionally, for the 10 L scale manufacturing process, this process improvement reduces the overall process time by 50%.
Downstream volume reduction and concentration
With demonstration of the XDR-10 scale-up, we turned our attention to downstream processing of the cells. In order to demonstrate a closed and automated solution, we subjected aliquots of 7 x 109 cells ( 2–3 L) of a 10 L expansion to processing through the Sefia™ Cell Processing System. We made an initial attempt to process PCS aggregates with the stock Sefia protocols. This was unsuccessful and resulted in clogging or fouling of the consumable kit, presumably due to the density differences between aggregates and single cells. Rather than exploring the operational space that may enable processing of cellular aggregates, we decided to process a 10 µM rho‐associated protein kinase inhibitor Y27632-mediated, single-cell solution and disaggregated with our passaging protocol as described. Originally designed and marketed for processing single-cell blood cell products, we demonstrated the utility of Sefia in processing PSCs. Run #1 reduced 2.0 L (7.39 x109 cells) to 240 mL in 1h 15min with 89% cell recovery (6.48 x109 cells) and 94% viability. Run #2 reduced 2.8 L (7.0 x109 cells) to 250 mL in 1h 27min with 79% recovery (5.55 x 109 cells) and 98% viability. These runs were not optimized in any way, and we feel recoveries could be further improved with additional replicates and fine-tuning of the processing protocol software by adjusting the operational parameters.
Conclusions
As regenerative medicine advances to clinical manufacturing, the need for closed, automated and scaled pluripotent stem cell manufacturing will increase. This work has presented process development and process improvements for a XDR-10 based, 10 L PSC manufacturing process, capable of producing >1010 cells per batch, with downstream concentration and volume reduction using the Sefia Cell Processing System.
We chose the hardware platforms with single-use, GMP manufacturing modality in mind. Since scalable stirred tank bioreactors from 0.1 L to 1 L are not commercially available, this meant choosing mixed hardware with differing configurations and properties. This presented some challenges to the direct application of traditional bioprocess engineering scale-up approaches. Choosing an appropriate small-volume, de-risking system was invaluable for process definition and examination of the unique sensitivities of culturing PSCs in stirred tank reactors, prior to attempting 10 L manufacturing runs.
Innovative solutions have been presented with implications for manufacturing, automation and process closure. The development of high-density seed banks offers many opportunities for process improvement, removing manual manipulation steps, shortening the expansion by 50% and decoupling subsequent suspension differentiation (Figure 7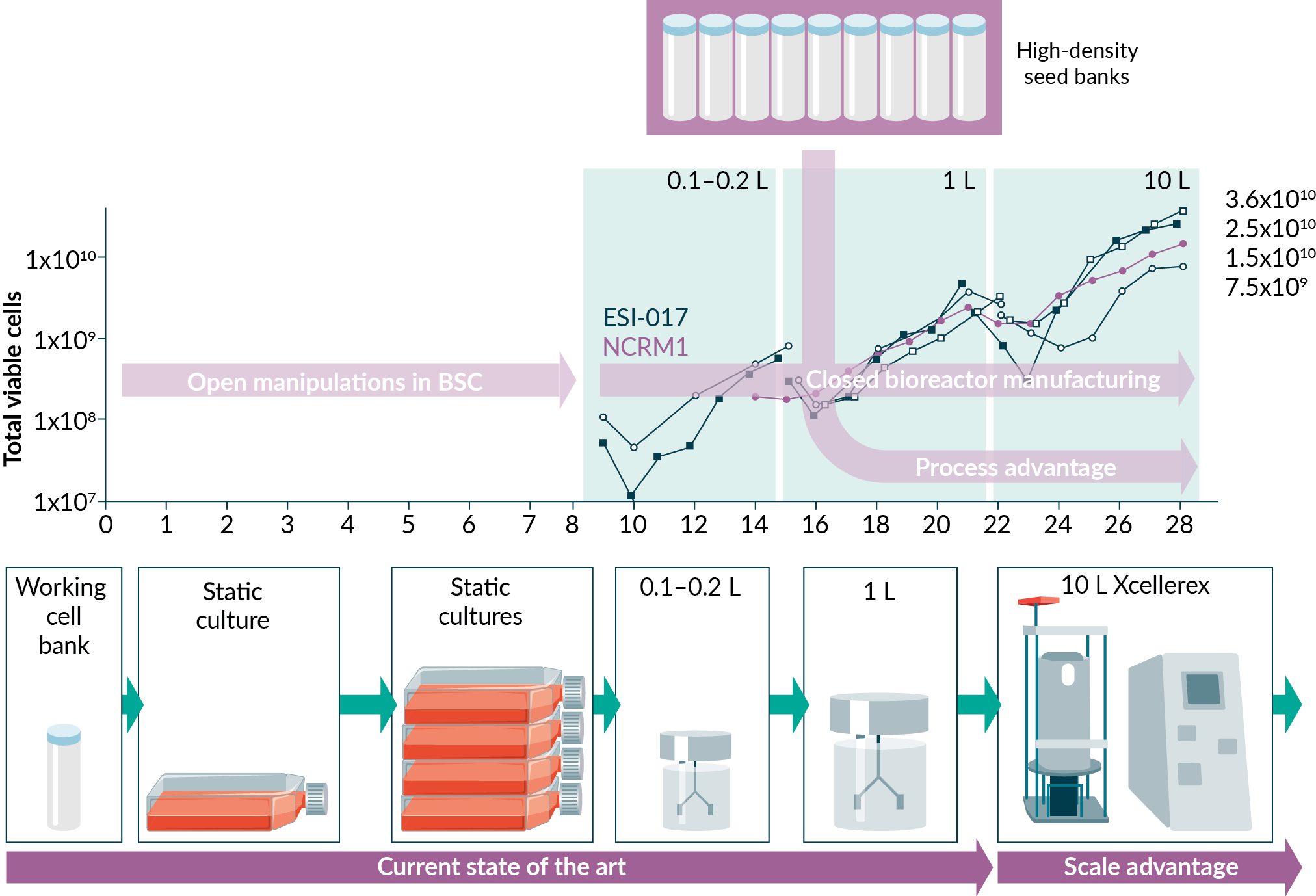
Taken together, this work demonstrates the relevant unit operations, process requirements and potential manufacturing improvements to set the stage for commercial production of PSCs. We expect this technology application will be further optimized and used to provide PSCs to differentiation workflows. Our hope is to enable the allogeneic cell therapy industry through facilitating the eventual clinical application of PSC-derived therapeutic cell types in advanced therapeutic products.
References
1. Amit M, Chebath J, Margulets V, Laevsky I et al. (2010). Suspension Culture of Undifferentiated Human Embryonic and Induced Pluripotent Stem Cells. Stem Cell Rev Rep. 2010; 6: 248–259. Crossref
2. Amit M, Laevsky I, Miropolsky Y et al. (2011). Dynamic suspension culture for scalable expansion of undifferentiated human pluripotent stem cells. Nat. Protoc. 2011; 6: 572. Crossref
3. Bareither R, Pollard D A review of advanced small-scale parallel bioreactor technology for accelerated process development: current state and future need. Biotechnol. Prog. 2011; 27, 2–14. Crossref
4. Carpenter M.K, Rao MS Concise Review: Making and Using Clinically Compliant Pluripotent Stem Cell Lines. Stem Cells Transl. Med. 2015; 4, 381–388. Crossref
5. Chen KG, Mallon BS, McKay RDG, Robey PG Human pluripotent stem cell culture: considerations for maintenance, expansion, and therapeutics. Cell Stem Cell 2014; 14, 13–26. Crossref
6. D’Amour KA, Bang AG, Eliazer S et al. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat. Biotechnol. 2006; 24, 1392–1401. Crossref
7. Hargus G, Cooper O, Deleidi M, et al. Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proc. Natl. Acad. Sci. 2010; 107, 15921–15926. Crossref
8. Hartman ME, Da DF, Laflamme MA Human pluripotent stem cells: Prospects and challenges as a source of cardiomyocytes for in vitro modeling and cell-based cardiac repair. Adv. Drug Deliv. 2016; Rev 96: 3–17. Crossref
9. Kempf H, Kropp C, Olmer R. Cardiac differentiation of human pluripotent stem cells in scalable suspension culture. Nat. Protoc. 2015; 10: 1345–1361. Crossref
10. Kroon E, Martinson LA, Kadoya K et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 2008; 26: 443–452. Crossref
11. Kropp C, Kempf H, Halloin C, et al. Impact of Feeding Strategies on the Scalable Expansion of Human Pluripotent Stem Cells in Single-Use Stirred Tank Bioreactors. Stem Cells Transl. Med. 2016; 5: 1289–1301. Crossref
12. Kwok CK, Ueda Y, Kadari A et al. Scalable stirred suspension culture for the generation of billions of human induced pluripotent stem cells using single-use bioreactors. J. Tissue Eng. Regen. Med. 2018; Crossref
13. Van Laake LW, Hassink R, Doevendans PA, Mummery C. Heart repair and stem cells. J. Physiol. 2006; 577, 467–478. Crossref
14. Li MD, Atkins H, Bubela T. The global landscape of stem cell clinical trials. Regen. Med. 2014; 9: 27–39. Crossref
15. Mohyeldin A, Garzón-Muvdi T, Quiñones-Hinojosa A (2010). Oxygen in Stem Cell Biology: A Critical Component of the Stem Cell Niche. Cell Stem Cell 2010; 7: 150–161. Crossref
16. Nienow AW, Coopman K, Heathman TRJ, Rafiq QA, Hewitt CJ (2016). Bioreactor Engineering Fundamentals for Stem Cell Manufacturing. In Stem Cell Manufacturing, J.M.S. Cabral, C.L. da Silva, L.G. Chase, and M.M. Diogo, eds. (Elsevier), 2016; 340. Crossref
17. Olmer R, Haase A, Merkert S, et al. Long term expansion of undifferentiated human iPS and ES cells in suspension culture using a defined medium. Stem Cell Res. 2010; 5: 51–64. Crossref
18. Olmer R, Lange A, Selzer S, et al. Suspension culture of human pluripotent stem cells in controlled, stirred bioreactors. Tissue Eng. Part C Methods 2012; 18: 772–784. Crossref
19. Phillips BW, Horne R, Lay TS, Rust WL, Teck TT, Crook JM. Attachment and growth of human embryonic stem cells on microcarriers. J. Biotechnol. 2008; 138: 24–32. Crossref
20. Pigeau G, Razvi A, Huang S. Predicting bioreactor product production based on independent or multivariate analysis of multiple physical attributes. 2018; US 62/665,155. Crossref
21. Pigeau GM, Csaszar E, Dulgar-Tulloch A. (2018). Commercial Scale Manufacturing of Allogeneic Cell Therapy. Front. Med. 2018; 5: 233. Crossref
22. Reubinoff BE, Pera MF, Fong CY, Trounson A, Bongso A. Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nat. Biotechnol. 2000; 18: 399–404. Crossref
23. Sackett SD, Brown ME, Tremmel DM, Ellis T, Burlingham WJ, Odorico JS. Modulation of Human Allogeneic and Syngeneic Pluripotent Stem Cells and Immunological Implications for Transplantation. Transplant. Rev. Orlando Fla 2016; 30: 61–70. Crossref
24. Simon MC, Keith B The role of oxygen availability in embryonic development and stem cell function. Nat. Rev. Mol. Cell Biol. 2008; 9: 285–296.
25. Takahash, K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007; 131: 861–872. Crossref
26. Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science 1998; 282: 1145–1147. Crossref
27. Trounson A, DeWitt ND. Pluripotent stem cells progressing to the clinic. Nat. Rev. Mol. Cell Biol. 2016; 17, 194–200. Crossref
28. Trounson A, Thakar RG, Lomax G, Gibbons D. (2011). Clinical trials for stem cell therapies. BMC Med. 2011; 9: 52. Crossref
29. Zweigerdt R. Large scale production of stem cells and their derivatives. Adv. Biochem. Eng. Biotechnol. 2006; 114: 201–235. Crossref
Affiliations
Shuohao Huang
Cytiva, Cell & Gene Therapy, Marlborough, MA, USA
Azher Razvi
Cytiva, Cell & Gene Therapy, Marlborough, MA, USA
Zoe Anderson-Jenkins
Centre for Commercialization of Regenerative Medicine, Toronto, ON, Canada
Danylo Sirskyj
Cytiva, Cell & Gene Therapy, Marlborough, MA, USA
Ming Gong
Centre for Commercialization of Regenerative Medicine, Toronto, ON, Canada
Anne-Marie Lavoie
Cytiva, Cell & Gene Therapy, Marlborough, MA, USA
Gary M Pigeau
Cytiva, Cell & Gene Therapy, Marlborough, MA, USA
Correspondence:
661 University Ave., Suite 1002
Toronto, ON, Canada
M5G 1M1
Tel.: +1 416 978 3751
gary.pigeau@cytiva.com
![]()
Authorship & Conflict of Interest
Contributions: All named authors take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Acknowledgements: The authors wish to acknowledge the Federal Economic Development Agency for Southern Ontario, GE Healthcare and CCRM for grant funding to establish the Centre for Advanced Therapeutic Cell Technologies. .
Disclosure and potential conflicts of interest: Dr Pigeau, Dr Huang and Dr Razvi have a patent Predicting Bioreactor Product Production Based On Independent Or Multivariate Analysis Of Multiple Physical Attributes pending.
Funding declaration: The authors received no financial support for the research, authorship and/or publication of this article.
Article & copyright information
Copyright: Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0 which allows anyone to copy, distribute, and transmit the article provided it is properly attributed in the manner specified below. No commercial use without permission.
Attribution: Copyright © 2020 Huang S, Razvi A, Anderson-Jenkins Z, Sirskyj D, Gong M, Lavoie AM, Pigeau GM. Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0.
Article source: Submitted; externally peer reviewed.
Revised manuscript received: Oct 28 2020; Publication date: Oct 22 2020.
- Results & Discussion
- Parameter optimization for a 10 L seed train
- Application of the optimized parameters to the expansion of a hiPSC line
- Correlation of hPSC growth kinetics with stirred tank bioreactorprocess parameters
- Scale-up of suspension aggregate culture to 1 L and 10 L reactors
- Scale-Up to the XDR-10
- High-density seed bank for direct inoculation of 1 L stirred tank bioreactor
- Downstream volume reduction and concentration
- Conclusions