
Promising non-viral vector for efficient and versatile delivery of mRNA for antigen-specific immunotherapy
Cell & Gene Therapy Insights 2020; 6(9), 1399–1409
10.18609/cgti.2020.154
Cellular immunotherapy involves the modification of immune cells in vivo or ex vivo to elicit, modulate or suppress immune responses. Modification by mRNA has become an attractive alternative to DNA vectors as a non-viral delivery approach, due to high transfection efficiency including in non-dividing cells. However, widespread adoption of this strategy is currently limited by the lack of commercially available ready-to-use reagents for in vivo delivery of mRNA. In this study, we evaluate the newly launched in vivo-jetRNA® for the delivery of mRNA-encoded antigens into nanoparticles and the resulting CD4+ T cell responses to one of the expressed epitopes. Four routes of administration were compared to determine the in vivo biodistribution of the nanoparticles based on evidence of antigen-specific T cell responses in various lymphoid tissues. Systemic routes achieved efficient delivery with most antigen-specific T cells stimulated in all sites tested. In the case of local routes, responses were confined to draining lymph nodes. Some of the activation markers (CD25, PD-1) were only induced in specific sites using specific routes, suggesting a role for the local dose of nanoparticles and the nature of antigen-presenting cells present involved in different sites. When applied to splenocytes from different mouse strains in vitro, the mRNA nanoparticles had marginal effect on the maturation of antigen-presenting cells, did not negatively affect viability and did not induce proinflammatory cytokines. We conclude that in vivo-jetRNA® is a promising mRNA formulation for efficient delivery of genes and antigens in vivo.
Cellular immunotherapy aims to harness the adaptive immune system in order to develop a targeted and curative response for unmet medical needs in the effective treatment of cancer, chronic infections or autoimmune diseases. The adaptive immune system is a specific immune response mainly driven by highly specialized and specific cells: lymphocytes (B and T). B lymphocytes, once exposed to an antigen in a peripheral lymphoid organ, can be activated and differentiate into plasma cells that secrete soluble or membrane-bound antigen-specific antibodies. T lymphocytes are subdivided into helper T cells (CD4+) and cytotoxic T cells (CD8+) whose role is to help activation of other immune cells (B cells, CD8+ T cells, macrophages, etc.) and to kill infected target cells, respectively. Successful immunogenic vaccines and therapies rests on activation of as many antigen-specific T cells as possible in different lymphoid tissues to create a robust immune response. Conversely, in the case of autoimmune diseases, effective immunotherapy relies on elimination or inactivation of self-reactive T cells. When nucleic acid vectors are used to express the antigen of interest or antigenic subparts (epitopes), they should be efficiently delivered to antigen-presenting cells (APCs) who will in turn present the encoded antigens to T cells in different lymphoid tissues. Thus, regardless of the desired outcome, there is a need for efficient and safe delivery modalities for successful antigen-specific and cellular immunotherapy. mRNA delivery on the rise
In vivo delivery of plasmid DNA to APCs was demonstrated to result in the induction of primary adaptive immune response more than two decades ago [1]Porgador A, Irvine KR, Iwasaki A, Barber BH, Restifo NP, Germain RN. Predominant role for directly transfected dendritic cells in antigen presentation to CD8+ T cells after gene gun immunization. J. Exp. Med. 1998; 188: 1075–82. . Since then, there has been a keen interest in DNA-based vaccines compared to protein/peptide-based vaccines; they are easier to produce, more cost-efficient and their delivery can be enhanced using transfection methods. Still, there are challenges with the use of plasmid DNA, mainly attributed to the insufficient immune response level elicited. This low immunogenicity of plasmid DNA is dependent on the delivery efficiency to target cells [2]Hobernik D, Bros, M. DNA vaccines - How far from clinical use? Int. J. Mol. Sci. 2018; 19(11): 3605. . To improve delivery of plasmid DNA into tissues, several delivery systems have been developed, such as cationic polymers, to condense and protect DNA from degradation while facilitating its delivery into APCs via endocytosis. As previously discussed [3]Nyamay’Antu A, Dumont M, Kedinger V, Erbacher P. Non-Viral vector mediated gene delivery: the outsider to watch out for in gene therapy. Cell Gene Ther. Insights 2019; 5: 51–7., we developed the leading cationic polymer-based transfection reagent in vivo-jetPEI® that is currently used in several ongoing nucleic acid-based drug development programs, with a majority of clinical trials for cancer treatment. As an alternative to DNA, mRNA-based immunotherapy is a promising tool due the fact that it retains many of DNA’s advantages including delivery of multiple antigens with one immunization while allowing a higher transfection efficiency, avoiding promoter-dependent inhibition of expression and genome integration [4]Kowalski PS, Rudra A, Miao L, Anderson DG. Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol. Ther. 2019; 27: 710–28. , [5]Liu MA. A Comparison of plasmid DNA and mRNA as vaccine technologies. Vaccines (Basel) 2019; 7(2): 37.. The higher transfection efficiency stems from the fact that mRNA does not need to reach the cell nucleus for expression nor require cell division for efficient gene expression. This is particularly interesting to improve the efficiency of delivery into slow-dividing and quiescent cells in vivo. The main drawback of mRNA is that it is not a stable molecule, hence the need for a transfection reagent that can efficiently protect mRNA during its in vivo delivery to cells and can easily be administered through systemic and local administration routes. The ideal transfection reagent should efficiently condense and protect mRNA from degradation by nucleases, as well as facilitate endosomal escape [6]Maruggi G, Zhang C, Li J, Ulmer JB, Yu D. mRNA as a transformative technology for vaccine development to control infectious diseases. Mol. Ther. 2019; 27: 757–72..
in vivo-jetRNA®, a promising vector for mRNA delivery
Delivery of mRNA into cells, and even more so in vivo is challenging because it depends on efficient condensation of mRNA molecules to prevent degradation by extracellular nucleases. Although significant progress has been made on efficient intracellular delivery of mRNA, thanks to extensive optimization efforts in the field to make mRNA-based therapeutic strategies viable for immunotherapy, there is still no such therapy that is approved and commercialized. Strategies for non-viral delivery of therapeutic mRNA include ex vivo[DCs as APCs)], [direct injection of naked mRNA], [in some cases], [specialized equipment], [for successful production. However]in vivo applications, we engineered a ready-to-use transfection reagent, in vivo-jetRNA®, specifically to ensure delivery of intact mRNA molecules to various tissues and cell types, the nature of which depending in part on the route of administration.
Case study: biodistribution, amplitude & quality of a
T cell response induced by an mRNA-encoded epitope delivered by in vivo-jetRNA®/mRNA nanoparticles
Delivery of antigen-encoding mRNA to exogenous DCs versus endogenous APCs
We have recently compared delivery of mRNA-encoded epitopes by ex vivo mRNA-electroporated DCs to nanoparticle delivery as means to elicit effective antigen-specific T cell responses in immunotherapy [7]Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.. We reported that nanoparticle-mediated mRNA delivery to professional (bone marrow-derived DCs) and non-professional (stromal) types of APCs in vitro using jetMESSENGER® was gentle and most efficient in stromal cells. While not designed for in vivo delivery, these mRNA-carrying nanoparticles achieved transfection of various hematopoietic APCs (conventional and plasmacytoid DCs and other myeloid cells; 1–7% of cells) and endothelial cells (
4% of cells) within lymph nodes. Using antigen-encoding mRNA, this level of transfection was adequate to induce robust CD4+ and CD8+ T cell responses, which showed a broader biodistribution than those induced by ex vivo mRNA-electroporated DCs injected by the same route (intraperitoneal). When administered intravenously, DCs were primarily retained in lungs, while the nanoparticles efficiently targeted lymphoid tissues such as the spleen. These studies took advantage of a new platform (Endotope) that enables optimal engagement of CD4+ and CD8+ T cells with multiple antigenic peptides encoded by DNA or mRNA [8]Dastagir SR, Postigo-Fernandez J, Xu C, Stoeckle JH, Firdessa-Fite R, Creusot RJ. Efficient presentation of multiple endogenous epitopes to both CD4+ and CD8+ diabetogenic T cells for tolerance. Mol. Ther. Methods Clin. Dev. 2016; 4: 27–3Dastagir SR, Postigo-Fernandez J, Xu C, Stoeckle JH, Firdessa-Fite R, Creusot RJ. Efficient presentation of multiple endogenous epitopes to both CD4+ and CD8+ diabetogenic T cells for tolerance. Mol. Ther. Methods Clin. Dev. 2016; 4: 27–3.
Systemic injection of in vivo-jetRNA®/mRNA nanoparticles induces robust & widespread antigen-specific T-cell responses
In this study, we evaluated how in vivo-jetRNA®, a specifically developed in vivo transfection reagent, performs in delivering mRNA-encoded epitopes to different lymphoid tissues based on route of administration. To this end, we assessed the amplitude and phenotype of antigen-specific T-cell responses elicited by in vivo[i.v.] and intraperitoneal [i.p.]) and two local routes (intradermal [i.d.] and subcutaneous [s.c.]) were compared(Figure 1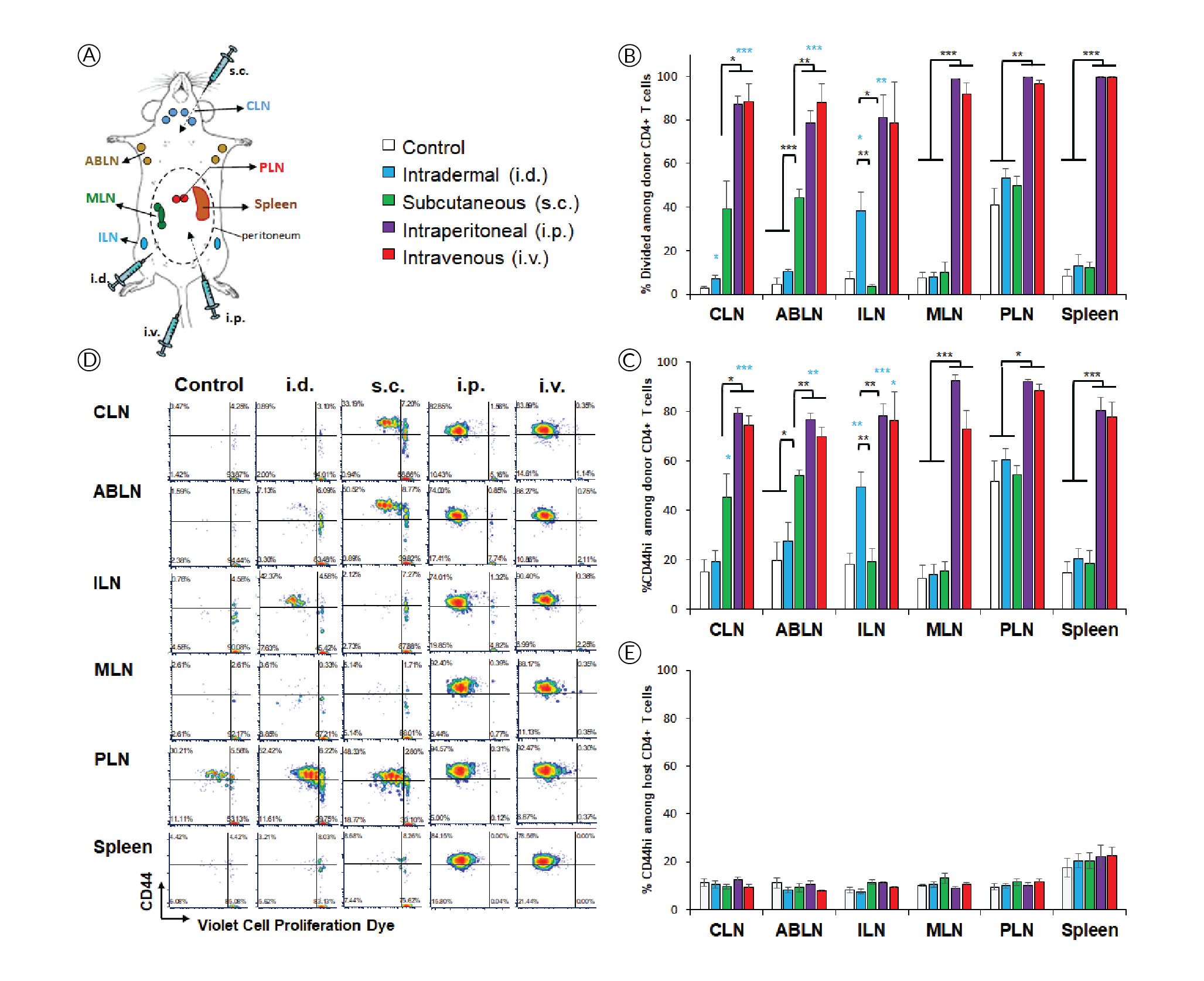
Local injection of in vivo-jetRNA® nanoparticles carrying antigen-encoding mRNA induce a T cell phenotype distinct from systemic administration
The high affinity interleukin-2 (IL-2) receptor chain CD25 can be induced upon activation and is important for T cell proliferation supported by IL-2 as the major T cell growth factor. However, it is interesting that, despite evidence of extensive proliferation (Figure 1), CD4+ T cells did not induce CD25 in response to systemic delivery (Figure 2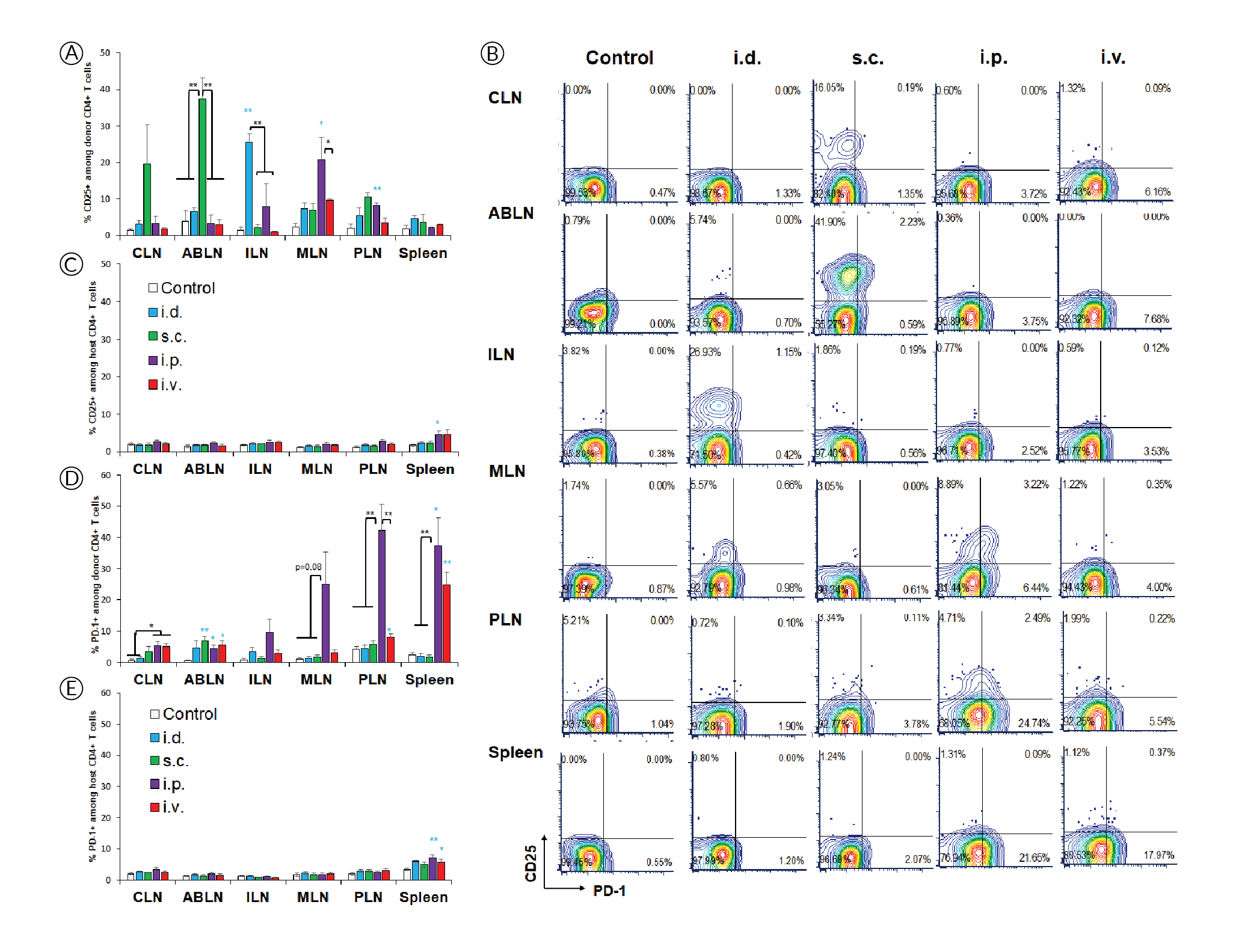
PD-1 is another activation marker that serves as negative regulator of the T cell response. PD-1 is usually more highly expressed on exhausted T cells following repeated antigen exposure. Interestingly, PD-1 upregulation was restricted to lymphoid tissues within the peritoneal cavity after i.p. injection (Figure 2B & D), contrasting with proliferation and CD44 upregulation (Figure 1B & C), which appear more sensitive. PD-1 was also prominently induced in the spleen after i.v. injection, which is where mRNA/nanoparticles primarily accumulate when using this delivery route [7]Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.. Again, the treatment did not affect PD-1 expression on other T cells (Figure 2E). Small but significant increases in CD25+ and PD-1+ polyclonal T cells were observed in spleen after systemic delivery.
The results of this study revealed interesting differences in the regulation of CD25 and PD-1 induction, which were uncoupled from simple TCR engagement that was evidenced by CD44 upregulation and followed by proliferation. Although more experimental work is needed for confirmation, our interpretation of those data is as follow:
- CD44 upregulation and proliferation in antigen-specific CD4+ T cells are achieved wherever there is sufficient antigen presentation. These responses are lower when antigen presentation is only contributed by migratory APCs (e.g. in draining lymph nodes after local delivery) and higher when resident APCs are directly transfected (e.g. systemic delivery). These responses are maximal when the local dose is higher (e.g. in peritoneal lymphoid tissues compared to skin-draining tissues after i.p. delivery).
- CD25 may be most readily induced by migratory APCs. In the case of i.d. and s.c. deliveries, these APCs may be dermal DCs and/or Langerhans cells from the skin. In the case of i.p. delivery, they may be APCs from the omentum, which serves as the port of entry for cells and particles introduced into the peritoneal cavity [9]Creusot RJ, Yaghoubi SS, Chang P, Chia J, Contag CH, Gambhir SS, Fathman CG. Lymphoid-tissue-specific homing of bone-marrow-derived dendritic cells. Blood 2009; 113(26): 6638–47., [10]Turley SJ, Lee JW, Dutton-Swain N, Mathis D, Benoist C. Endocrine self and gut non-self intersect in the pancreatic lymph nodes. Proc. Natl. Acad. Sci. USA 2005; 102(49): 17729–33.Turley SJ, Lee JW, Dutton-Swain N, Mathis D, Benoist C. Endocrine self and gut non-self intersect in the pancreatic lymph nodes. Proc. Natl. Acad. Sci. USA 2005; 102(49): 17729–33..
- In contrast, PD-1 may be more readily induced by resident APCs, but at a higher local dose than is required for CD44 upregulation and proliferation, thus is not observed in distal lymph nodes. Although these responses are prominent in all lymphoid tissues after i.p. injection for example, we find that they are nonetheless significantly lower (p<0.03) in ABLN and CLN (non-peritoneal) than in MLN and PLN (peritoneal). The same may apply for the spleen as it is within the peritoneal cavity. Nanoparticles that are injected i.v. disperse easily and can reach all lymphoid tissues with a dose sufficient for CD44 upregulation and T cell proliferation, but not necessarily for PD-1 upregulation. However, the spleen is by far the most vascularized of the tissues evaluated, and as such, it is expected to have a higher local dose of nanoparticles [7]Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62., thereby enabling PD-1 upregulation. The small effect on CD25 and PD-1 seen on splenic polyclonal CD4+ T cells may be attributed to this higher accumulation of nanoparticles combined with cumulated T cell responses to other epitopes, besides p79, expressed by the mRNA.
The differences seen between routes may reflect both local antigen doses and the nature of APCs transfected and/or presenting antigens, keeping in mind that transfected cells incapable of presenting antigens may nonetheless transfer encoded antigens to professional APCs. The environment of each lymphoid tissue may also influence the immune responses. Both MLN and PLN drain the intestinal mucosa [10]Turley SJ, Lee JW, Dutton-Swain N, Mathis D, Benoist C. Endocrine self and gut non-self intersect in the pancreatic lymph nodes. Proc. Natl. Acad. Sci. USA 2005; 102(49): 17729–33.Turley SJ, Lee JW, Dutton-Swain N, Mathis D, Benoist C. Endocrine self and gut non-self intersect in the pancreatic lymph nodes. Proc. Natl. Acad. Sci. USA 2005; 102(49): 17729–33., and their APC content and microenvironment milieu is expected to differ dramatically from skin-draining lymph nodes (ABLN, CLN, ILN). Substantial heterogeneity in immune responses is even observed within gut-draining lymph nodes depending on the sections of the gut they drain [11]Esterházy D, Canesso MCC, Mesin L, Muller PA, de Castro TBR, Lockhart A, ElJalby M, Faria AMC, Mucida D. Compartmentalized gut lymph node drainage dictates adaptive immune responses. Nature 2019; 569(7754): 126–30.. Thus, it is important to understand the biodistribution of T cell responses and how it affects the profile of the responding T cells, and select the route accordingly.
in vivo-jetRNA® nanoparticles do not exhibit toxicity & overstimulation of proinflammatory cytokines on immune cells in vitro
Since in vivo-jetRNA® is a recently launched formulation for mRNA delivery, we addressed whether mRNA formulated with this reagent had deleterious effects on immune cells, including an excessive non-specific release of proinflammatory cytokines by overstimulation of Toll-like receptors (TLR). To gain insight of these possible effects on immune cells across genetically diverse individuals, we cultured splenocytes from three strains of mice for 24h in the presence of ‘naked’ mRNA, mRNA/in vivo-jetRNA® nanoparticles or a variety of TLR ligands. We used a relatively high dose of mRNA (0.5 μg per 2x105 cells, same mRNA as in vivo studies), which is only ten times lower than the dose injected in vivo, and we used comparable amounts of TLR ligands. The mRNA nanoparticles had no to marginal effects in increasing the expression of MHC and costimulatory molecules on the surface of CD11c+ cells (Figure 3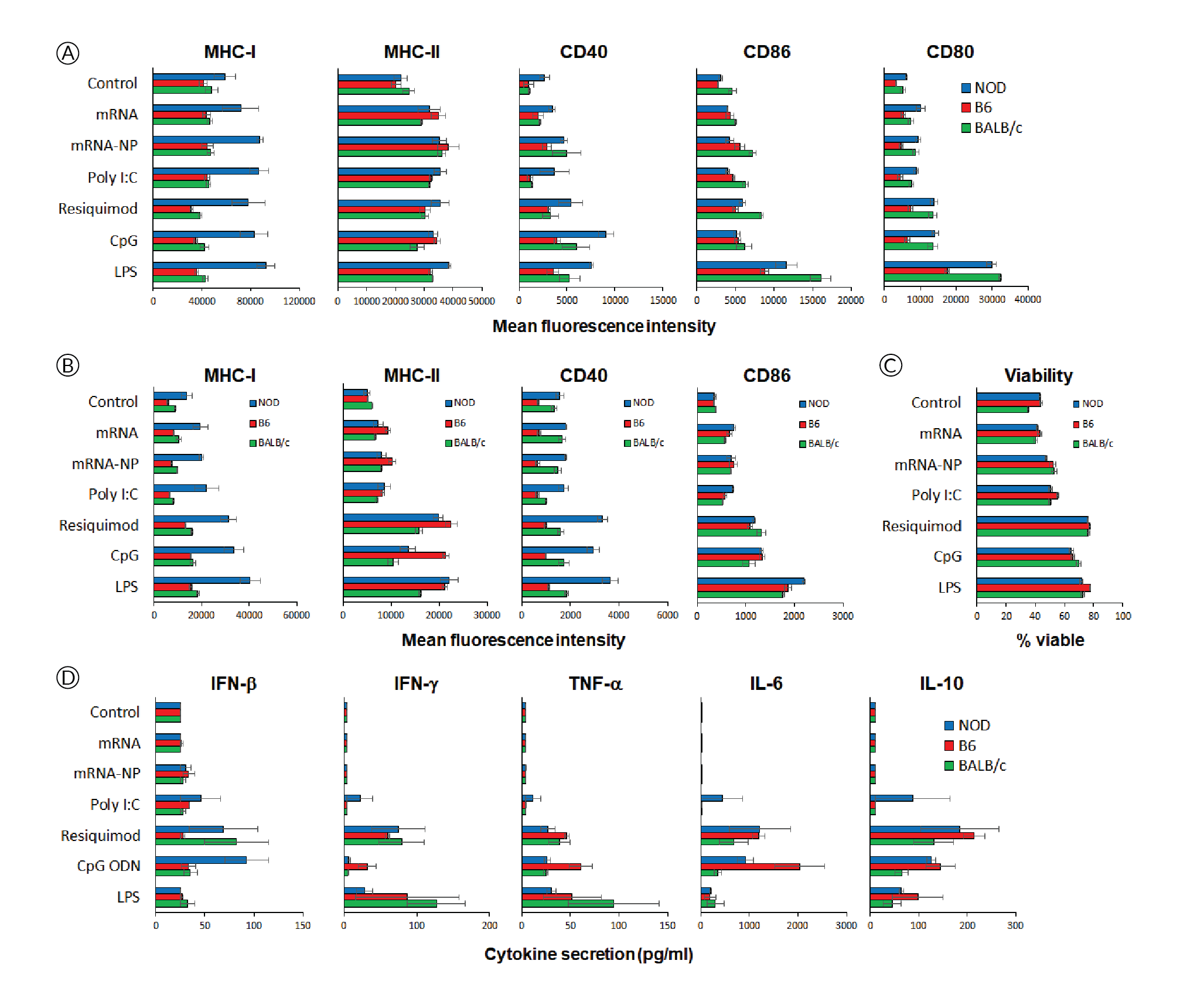
in vivo-jetRNA® constitutes a versatile delivery vehicle for
mRNA-encoded antigens
Irrespective of the administration route used, systemic or local, this data set shows that in vivo-jetRNA® is a promising non-viral delivery modality for effective in vivo delivery of therapeutic mRNA. Our previous studies demonstrated that the delivery of mRNA using nanoparticles can successfully target antigens to various types of APCs in several lymphoid tissues [7]Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62., a winning combo to elicit a robust immune response. The phenotype and expansion of the stimulated T cells can be modulated based on the route of delivery, suggesting that this approach is versatile. The relative contribution of different transfected APCs to the overall immune response and how route of vector delivery, microenvironment of the target lymphoid tissue and dose, as parameters, modulate the quality of the T cell response remain to be further explored using models in which the outcome of the T cell response can be tested (viral clearance, tumor rejection, etc.).
Methods
NOD mice (strain #001976, females, 8 weeks of age), used as recipients, and T cell receptor-transgenic congenic (CD45.2) BDC2.5 mice (cross of strains #004460 and #014149, females, 8–16 weeks of age), used as donors, were obtained from The Jackson Laboratory, the latter bred in our barrier facility. All procedures were performed following protocols approved by the Columbia University Institutional Animal Care and Use Committee. In vitro-transcribed mRNAs, expressing multiple epitopes including the p79 mimotope recognized by BDC2.5 T cells[7]Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62.Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62., [8]Dastagir SR, Postigo-Fernandez J, Xu C, Stoeckle JH, Firdessa-Fite R, Creusot RJ. Efficient presentation of multiple endogenous epitopes to both CD4+ and CD8+ diabetogenic T cells for tolerance. Mol. Ther. Methods Clin. Dev. 2016; 4: 27–3Dastagir SR, Postigo-Fernandez J, Xu C, Stoeckle JH, Firdessa-Fite R, Creusot RJ. Efficient presentation of multiple endogenous epitopes to both CD4+ and CD8+ diabetogenic T cells for tolerance. Mol. Ther. Methods Clin. Dev. 2016; 4: 27–3, was produced by TriLink Biotechnologies. The mRNA was produced with modifications (Anti-Reverse Cap Analog as well as 5-methyl-cytosine and pseudouridine substitutions) to increase stability and reduce immunogenicity. For isolation of antigen-specific CD4+ T cells, spleen and pooled lymph nodes were collected from donor CD45.2+ BDC2.5 mice and CD4+ CD25- T cells were purified using the MoJo™ Mouse CD4 T Cell Isolation Kit (BioLegend) supplemented with biotinylated anti-CD25. Cells were then labelled with Violet Cell Proliferation Dye (eBioscience) and 0.5–1x106 T cells were injected i.v. into recipient NOD (CD45.1+) mice. The formulation of mRNA into in vivo-jetRNA®/mRNA nanoparticles was done according to manufacturer’s instructions. Following adoptive transfer, recipient mice were injected with nanoparticles containing 5 μg mRNA per mouse either i.v. (tail vein), i.p., i.d. (shaved abdominal area) or s.c. (neck fold) (Figure 1A). Control mice received T cells but no nanoparticle treatment. After 3 days, lymphoid tissues (as shown in Figure 1A) were collected separately and processed for single-cell suspensions. The cells were stained with antibodies to CD4, CD45.2, CD25, CD44 and CD279 (PD-1), all from BioLegend, and analyzed on a BD Fortessa™ flow cytometer. Splenocytes from NOD, C57BL/6 (B6) and BALB/c mice were cultured in vitro for 24h in the presence of mRNA with or without formulation with in vivo-jetRNA® or with various TLR ligands known to stimulate antigen-presenting cell maturation and cytokine secretion. Supernatant from these cultures were separately analyzed for 13 cytokines using the LegendPlex™ Mouse Inflammation Panel (BioLegend). The cells from these cultures were analyzed on a BD Fortessa™ flow cytometer after staining for B220 (B cells), CD11c (primarily dendritic cells), MHC class I (H2-Kd for NOD and BALB/c, H2-Kb for B6), MHC class II (I-A/I-E for B6 and BALB/c, I-Ak for NOD), CD40, CD80, CD86 as well as propidium iodide (all reagents from BioLegend and BD Bioscience). Flow cytometry data were analyzed with FCS Express 7.
References
1. Porgador A, Irvine KR, Iwasaki A, Barber BH, Restifo NP, Germain RN. Predominant role for directly transfected dendritic cells in antigen presentation to CD8+ T cells after gene gun immunization. J. Exp. Med. 1998; 188: 1075–82. Crossref
2. Hobernik D, Bros, M. DNA vaccines - How far from clinical use? Int. J. Mol. Sci. 2018; 19(11): 3605. Crossref
3. Nyamay’Antu A, Dumont M, Kedinger V, Erbacher P. Non-Viral vector mediated gene delivery: the outsider to watch out for in gene therapy. Cell Gene Ther. Insights 2019; 5: 51–7. Crossref
4. Kowalski PS, Rudra A, Miao L, Anderson DG. Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol. Ther. 2019; 27: 710–28. Crossref
5. Liu MA. A Comparison of plasmid DNA and mRNA as vaccine technologies. Vaccines (Basel) 2019; 7(2): 37. Crossref
6. Maruggi G, Zhang C, Li J, Ulmer JB, Yu D. mRNA as a transformative technology for vaccine development to control infectious diseases. Mol. Ther. 2019; 27: 757–72. Crossref
7. Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol. Ther. Methods Clin. Dev. 2020; 16: 50–62. Crossref
8. Dastagir SR, Postigo-Fernandez J, Xu C, Stoeckle JH, Firdessa-Fite R, Creusot RJ. Efficient presentation of multiple endogenous epitopes to both CD4+ and CD8+ diabetogenic T cells for tolerance. Mol. Ther. Methods Clin. Dev. 2016; 4: 27–38. Crossref
9. Creusot RJ, Yaghoubi SS, Chang P, Chia J, Contag CH, Gambhir SS, Fathman CG. Lymphoid-tissue-specific homing of bone-marrow-derived dendritic cells. Blood 2009; 113(26): 6638–47. Crossref
10. Turley SJ, Lee JW, Dutton-Swain N, Mathis D, Benoist C. Endocrine self and gut non-self intersect in the pancreatic lymph nodes. Proc. Natl. Acad. Sci. USA 2005; 102(49): 17729–33. Crossref
11. Esterházy D, Canesso MCC, Mesin L, Muller PA, de Castro TBR, Lockhart A, ElJalby M, Faria AMC, Mucida D. Compartmentalized gut lymph node drainage dictates adaptive immune responses. Nature 2019; 569(7754): 126–30. Crossref
Affiliations
Rebuma Firdessa-Fite
Columbia Center for Translational Immunology, Columbia University Irving Medical Center, New York, NY, USA
Jorge Postigo-Fernandez
Columbia Center for Translational Immunology, Columbia University Irving Medical Center, New York, NY, USA
Valérie Toussaint-Moreau
Polyplus-transfection, Illkirch, France
Fabrice Stock
Polyplus-transfection, Illkirch, France
Alengo Nyamay’antu
Polyplus-transfection, Illkirch, France
Patrick Erbacher
Polyplus-transfection, Illkirch, France
Rémi J Creusot
Columbia Center for Translational Immunology, Columbia University Irving Medical Center, New York, NY, USA

Authorship & Conflict of Interest
Contributions: Rebuma Firdessa-Fite made all mRNA nanoparticle preparations, performed in vivo experiments and multiplex cytokine assays, Jorge Postigo-Fernandez set up the in vitro treatment of splenocytes and performed their flow cytometric analysis, Valérie Toussaint-Moreau, Fabrice Stock and Patrick Erbacher worked on the development of in vivo-jetRNA®, Patrick Erbacher and Rémi J Creusot designed and directed the study, Alengo Nyamay’antu and Rémi J Creusot wrote the manuscript.
Acknowledgements: None.
Disclosure and potential conflicts of interest: Rémi J Creusot is the inventor of the Endotope platform for optimal presentation of nucleic acid-encoded epitopes (US20170283810).
Funding declaration: Rebuma Firdessa-Fite and Jorge Postigo-Fernandez are funded by fellowships from the American Diabetes Association (1-19-PMF-022 and 1-18-PDF-151, respectively). The in vitro study was supported by the Translational Therapeutics Accelerator program funded by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant Number UL1TR001873.
Article & copyright information
Copyright: Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0 which allows anyone to copy, distribute, and transmit the article provided it is properly attributed in the manner specified below. No commercial use without permission.
Attribution: Copyright © 2020 Firdessa-Fite R, Postigo-Fernandez J, Toussaint-Moreau V, Stock F, Nyamay’antu A, Erbacher P & Creusot RJ. Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0.
Article source: Invited.
Revised manuscript received: Oct 26 2020; Publication date: Oct 29 2020.