Making an efficient & effective transition to GMP raw materials
Cell & Gene Therapy Insights 2019; 5(2), 239-248.
10.18609/cgti.2019.028

Q What challenges can occur in switching from preclinical to GMP grade raw materials at a late stage, and can they be quantified?
UN: One of the challenges on the way to commercialization is the transition from preclinical development to the clinical stage. This transition involves, amongst others, key considerations regarding the raw materials that are used in the manufacturing process. In the initial preclinical phase the safety and quality of raw materials often have a lower priority since these are not intended to be used for clinical administration. At a later stage, during the late clinical phases and commercial production it is crucial to use safe and high quality raw materials in the ATMP manufacturing process. In addition, many health authorities and the EU-GMP Guideline (EudraLex Volume 4, Part 4) recommend to use the highest grade raw materials available.
Changing raw materials at a late stage in clinical development often creates significant additional costs and loss of precious time. These additional costs are only to a small extent caused by slightly higher costs of GMP grade raw materials. They are primarily driven by the need to perform time-consuming clinical comparability studies to prove the raw material changes do not alter the final ATMP. Switching to the required GMP grade raw materials at an early stage, i.e. directly after preclinical development, will prevent the need for such studies and thereby brings a significant economic benefit. A study performed by the Tufts Center for the Study of Drug Development (CSDD) estimated that the costs of an amendment for a phase III trial costs more than three times as much as an amendment for a phase II trial . They estimated the average direct cost to make changes to the protocols of phase III trials tops $1 million per study.
Q What are the key preparatory steps in preclinical development to enable a smooth transition to GMP in terms of raw materials and suppliers?
UN: To enable a seamless transition from preclinical development to the clinical phases it is recommended to identify the appropriate GMP grade raw materials and reliable suppliers already during early stage preclinical research and prior to clinical development of the ATMP.
Points to be considered:
- Safety:
Since raw materials are in direct contact with cells that are intended for clinical administration, the safety and quality of the raw material used will affect the potency and safety of the final cell product. Therefore, raw materials have to be qualified to be suitable for their intended use and have to be safe. To mitigate and control the risk of adventitious agents, as potential contaminants in raw materials of biological origin, they should be serum-free and free of animal derived components. A supplier of raw materials should have an animal-derived component-free and serum-free policy in place and ensure its compliance with these policies. - Regulatory and GMP Compliance:
A basic requirement for manufacturing GMP grade raw materials is a robust and well-maintained quality management system (QMS) to ensure suitable processes are in place and executed as defined. GMP products should be manufactured, tested, released and distributed under an ISO 9001:2015 certified Quality Management System.
Raw materials have to be consistently manufactured to a pre-defined quality standard. Using such raw materials has a positive impact on the consistency of the ATMP manufacturing process. In order to obtain such consistent quality of the raw materials, production and quality control should follow all applicable GMP standards with well controlled processes in place. Since there are no legal requirements for raw materials to be manufactured within a particular quality system and raw material suppliers claim a self-committed GMP compliance, it is important for the ATMP manufacturer to understand the interpretation of GMP standards of the different suppliers. To conduct supplier audits is a good practise to confirm the quality management system and GMP standards are appropriate and applied as required.
In addition, compliance with quality regulations as defined in the USP Chapter <1043> and Ph. Eur. General Chapter 5.2.12 have to be considered and GMP cytokines should be tested and released according to USP Chapter <92> as applicable. - Regulatory Support:
Regulatory support during the clinical phases is an important success factor. The supplier of GMP grade raw materials should be capable of providing expert regulatory and technical support as well as FDA Drug Master Files for the raw materials. Customized solutions can meet special compliance needs. An important prerequisite is a transparent communication between the ATMP manufacturer and the supplier. This includes information on how a supplier mitigates the risk of contamination with adventitious agents.
Q What factors do you see as critical to ensuring sufficient supply of materials at late-phase clinical and commercial scale?
UN: It is not sufficient to identify a supplier that can provide a raw material in the required quality. The supply chain security and consistency of quality must also be considered as it is an important factor for successful commercialization of ATMPs. Part of a secure supply chain is the timely and reliable availability of raw materials with consistent product quality. A delay in delivery or change in product quality could lead to delays in production, not only increasing costs but also putting precious patient samples in jeopardy.
As part of a supply chain management system the supplier should have different quality and security measures in place such as:
- Following the relevant Good Distribution Practice (GDP) guidelines
- Authorized Economic Operator (AEO) status
- Dedicated stability studies to confirm product quality during shipment
- Have supply agreements in place
- Reserve batches
- Capacity to manufacture large quantities of raw material
- Clear and transparent communication of time lines
Q Cell & gene therapy abounds with highly specialised materials – how can one ensure a robust supply of these as and when they are required?
UN: When scaling up from clinical trials in phase I to large-scale commercial manufacturing the number of patients (i.e. doses to be produced) might go up by several magnitudes. As a consequence, the supply of GMP grade raw materials is getting even more critical. The safest way to eliminate possible problems with the supply of GMP grade raw materials is by having a supply agreement in place well before large-scale manufacturing is planned to commence. Giving the supplier a clear and reliable forecast of doses to be produced and consequently the amounts of raw material needed, will allow the supplier to produce large size batches for a dedicated customer and have enough material available on demand.
To be able to provide high quality raw materials at strongly increasing quantities the supplier needs to have the flexibility to scale-up the manufacturing capacities. It will also be an advantage to keep a stock of material close to the market of high demand.
Q What quality standards do CellGenix materials meet in order to satisfy GMP regulations on a global basis?
UN: To prevent deviations in a cell therapy manufacturing process it is crucial to choose raw materials with a consistent high product quality.
CellGenix is capable of manufacturing a product with a consistently high quality, as all our preclinical and GMP grade products are manufactured, tested, released and distributed under an ISO 9001:2015 certified Quality Management System (QMS), and all globally relevant GMP regulations are followed for GMP grade products.
Internal quality audits are performed periodically by qualified CellGenix staff, and customers conduct audits on a regular basis, which gives us the opportunity to continuously improve our QMS.
Having a supplier qualification program in place is essential to secure the entire supply chain. CellGenix qualifies and performs audits of all suppliers for critical components prior to allowing these materials to be used in the manufacturing process.
Further processes are established at CellGenix to continuously improve the quality system. The complete manufacturing process up to the final product is closely monitored and the feedback from the market is also taken into account for further improvements. Whenever needed to improve the quality system Corrective and Preventive Actions (CAPA) are executed.
For full transparency and to qualify CellGenix as a supplier of GMP grade raw materials, which mitigates the risk to an acceptable level, we provide
- Detailed batch specific test results on a Certificate of Analysis (CoA)
- The possibility to audit the manufacturing site
- Comprehensive product-specific documentation (e.g. Regulatory Support Files, TSE Certificates, Drug Master Files (DMFs))
- Technotes on product stability and consistency studies performed by QC
Q What role does CellGenix play in ongoing standards development for raw materials?
UN: The manufacturing of raw materials used in cell and gene therapy manufacturing is not well regulated and not supervised by any health authority. There were some guidelines issued for ATMP manufacturers, with some regulations on raw materials, but in our opinion, most of those guidelines do not offer the level of guidance needed. With USP Chapter <1043>, the FDA only offers a recommendation for raw material users and not for the manufacturers of raw materials. To help improve regulatory guidance we are actively involved in many of the regulatory initiatives and discussions. Together with the USP we have written the first version of USP chapter <92> for “Growth Factors and Cytokines Used in Cell Therapy Manufacturing” and were actively involved in the discussions for the setup of Ph. Eur. General Chapter 5.2.12. This chapter is referred to in the EU-GMP-guideline EudraLex Volume 4 Part 4, and therefore gets a binding character.
With the participation of CellGenix a new International Technical Standard (ISO/TS-20399) was released by the International Organization of Standardization (ISO) in November 2018 providing guidance for raw material (“Ancillary Materials”) suppliers.
Q Why is implementing an ADC- and serum-free policy so important in relation to cytokines and media in particular?
UN: Both, cytokines and cell culture media are used as raw materials in ATMP manufacturing processes and are not intended to be part of the final product. Nevertheless, there is always the potential risk of contaminating the cell and gene therapy product with adventitious agents such as bacteria or viruses, which pose a risk to patients’ safety.
Therefore, it is important to have a strict animal-derived component-free (ADCF) policy in place and avoid where feasible animal- or human-derived components in the production to ensure maximum safety of the preclinical and GMP raw materials.
Media should be produced without the addition of human or animal serum in order to minimize the risk of contaminations with adventitious agents. Nevertheless, some of the media products may still contain animal- or human-derived components, but only material is accepted if it represents no apparent health hazard and is compliant with the current TSE guidelines (EMEA/410/01), ICH Q5A and EP chapter 5.1.7, as applicable.
Q And how do you ensure this policy is strictly adhered to?
UN: The large majority of our cytokines is produced in our dedicated animal-free facility (ADCF Level 2). These cytokines have never been exposed to animal components or by-products. They can therefore be safely used without the need to perform time-consuming and expensive viral safety studies, thereby bringing a significant economic benefit. Those very few cytokines produced in our segregated animal-derived components (ADC) production area require either the use of ADC in the production process or a human expression system to allow proper functional expression (ADCF Level 1), but the product itself does not contain any ADC.
Our cytokine product portfolio is divided into two distinct ADCF levels (Figure 1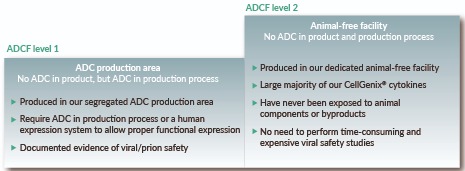
All materials used in the production of raw materials are formally approved by the Quality Unit. As part of the material control, they are procured from reliable manufacturers and suppliers and their origin and impurity profile are assessed before use. The safety of the raw material is demonstrated by certificates of origin and validation reports. In addition, TSE certificates are provided for all our cytokines.
In order to facilitate a risk assessment performed by our customers we assess each material used for production of our cytokines for the absence of ADC in its manufacturing process. Whenever possible, materials are used that themselves were produced without the use of ADC.
For the few media that still contain animal- or human-derived components, we are continuously evaluating and assessing to replace those components with materials of non-animal and non-human origin.
If human, but no animal-derived materials are part of the media formulation, the product is designated “xeno-free”. Human proteins that are part of the media formulation have been collected from healthy donors at the time of collection. All samples were tested individually and found negative for viral diseases by well-established methods.
Q What role can raw materials suppliers play in helping to ensure batch-to-batch consistency in cell & gene therapy manufacture?
UN: As mentioned before and stated in the EU-GMP guideline, particular attention should be paid to minimizing as much as possible the variability of the quality of the raw material, since it affects the quality, safety and efficacy of the advanced therapy medicinal products (ATMPs). Therefore, it is crucial that the supplier provides raw material to the ATMP manufacturer with a highly consistent, predefined product quality.
Inconsistent product performance of raw materials causes deviations in the cell therapy manufacturing process. It is unlikely that the ATMP manufacturer will be able to use the same batch of raw material during all stages from early clinical studies to commercial manufacturing. It is therefore crucial to choose GMP raw materials with a high and reliable batch-to-batch consistency. This will allow the ATMP manufacturer to establish the manufacturing process as accurately as possible and save time and cost of goods on incoming controls and re-validations.
To make sure a manufacturing process is not influenced by using different batches of GMP cytokines, CellGenix has performed batch-to-batch consistency studies for its key CellGenix® GMP cytokines. These studies demonstrate that our batch-to-batch consistency is very high over the full range of production.
Batch-to-batch consistency of critical raw materials is currently not emphasized in most of the different regulatory guidelines. It is now defined in the ISO Technical Standard 20399: Ancillary materials present during the production of cellular therapeutic products, issued in November 2018.
Q Finally, 2019 marks the 25th birthday of CellGenix – can you reflect on the changes wrought in cell & gene therapy over this period, and what trends you expect to see in the coming years?
UN: I have been with CellGenix since early 2018. Already during this time, the cell & gene therapy industry has changed a lot. Having the 25th birthday of CellGenix is a big success story not only for the company itself but also for the field of cell & gene therapy. The company was founded in 1994 as a spin-off of the Freiburg University Medical Center and was the first European company to obtain a GMP manufacturing license for cell processing in 1995.
During the past 25 years a lot has changed at CellGenix and in the industry. Regulatory frameworks for manufacturing and market approval of ATMPs have been established, which also concern the raw materials used for further processing of these ATMPs. Important to note is that an international harmonization for specific regulatory topics is on the way which hopefully will lead to smoother processes.
Great progress has been achieved in the science behind cell & gene therapies. Scientists have a better understanding of the diseases but also on how to grow, activate, differentiate and genetically alter cells in vitro. In addition to that, new tools and technologies, like CRISPR /Cas9 or the invention of iPSCs, as well as better analytical methods have emerged. Furthermore, significant clinical progress has been demonstrated. Inherited diseases like ADA-SCID or hemophilia can be successfully treated. Also, the first CAR-T therapies received marketing authorization, which can be seen as the next big step to cure certain types of cancer.
The part of your question related to trends is not easy to answer as the future is hard to predict. Nevertheless, there are some things where I expect progress. I believe that cell and gene therapies such as CAR-T will improve their safety and efficacy as more and more data will be available. Some of the therapies might also be used for solid cancers in the future. I also believe that automation, scale-up effects and closed systems will have an important role to play in the facilitation of ATMP manufacturing. This will lead to a reduction of cost of gods while still maintaining a high quality. Furthermore, I believe that new and innovative reimbursement models will emerge to make these therapies available for more and more patients.
Affiliation
Dr Uno Nirenberg
CellGenix



This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.