When using a closed and automated manufacturing platform, is there an option to maintain flexibility?
Cell & Gene Therapy Insights 2021; 7(7), 857–869
10.18609/cgti.2021.117
To allow greater flexibility and better targeting of CAR T cell therapies, Astellas developed convertibleCAR™ T cells, which kill antigen-expressing target cells only in the presence of an activating bispecific adapter. Here, we will discuss the clinical manufacturing of autologous convertibleCAR T cells, with a special focus on automation of the process using the Lonza Cocoon® Platform.
The Astellas convertibleCAR platform
Kaman Kim
The pioneering CAR T therapies initially approved for clinical use are technically limited. They have a single-purpose scFv receptor, no way to control the dose of any given cell, and no mechanism to address tumor heterogeneity or antigen loss. All of these issues can cause CAR T therapies to fail – either during treatment or due to relapse and antigen loss after administration.
To address these limitations, Xyphos Biosciences – an Astellas company – created a universal chimeric antigen receptor (CAR; Figure 1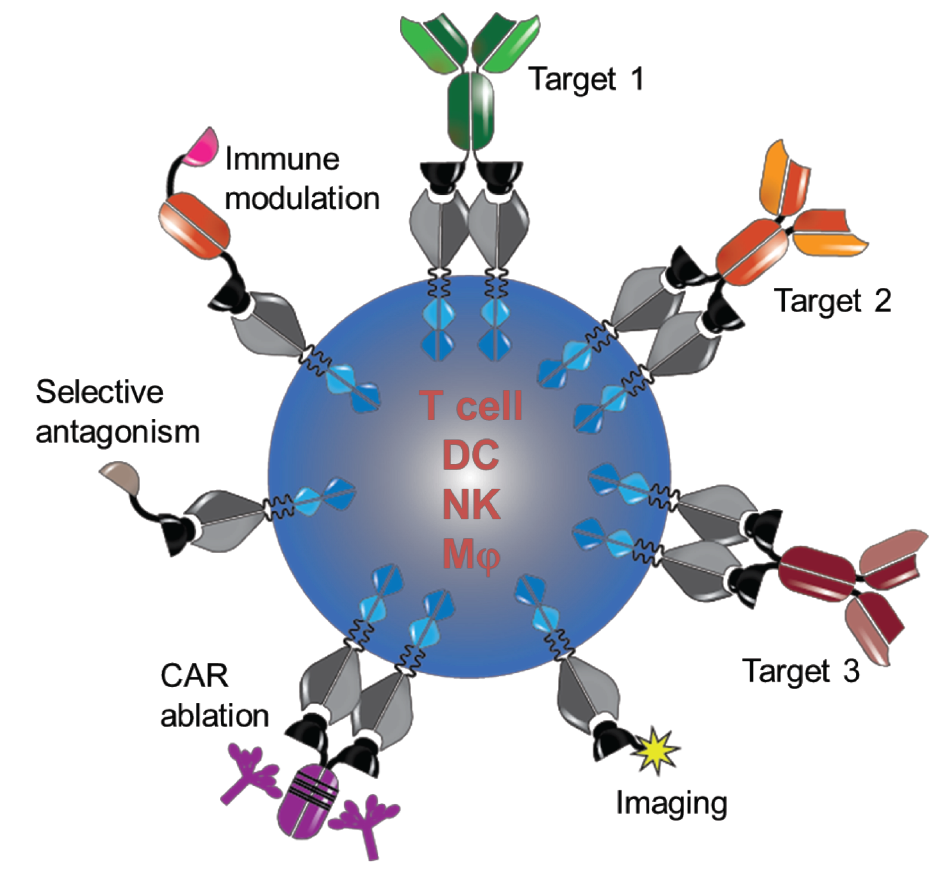
Our platform provides a multifunctional receptor, addressing the single-purpose limitation of an scFv CAR and allowing for easy retargeting of our chimeric antigen-bearing cell to any tumor target of interest using a bispecific adaptor molecule. We are also able to control the function of the CAR T cell by changing the ratios of adaptor molecules as needed. Lastly, by combining receptors we can multiplex and target multiple antigens simultaneously.
Developing the platform
Our starting point was the NKG2D-MIC signaling axis. NKG2D is an activating receptor that is present on natural killer (NK) cells and allows them to survey the body for cells that are stressed, often due to viral or oncogenic transformation. Stressed cells upregulate MIC ligands on the cell surface, which bind to NKG2D receptors on NK cells and prompt them to eliminate the unhealthy cell.
Applying a structure-based engineering approach, we mutated the extracellular domain of NKG2D to render the receptor inert (iNKG2D) and incapable of binding to any natural human ligands (Figure 2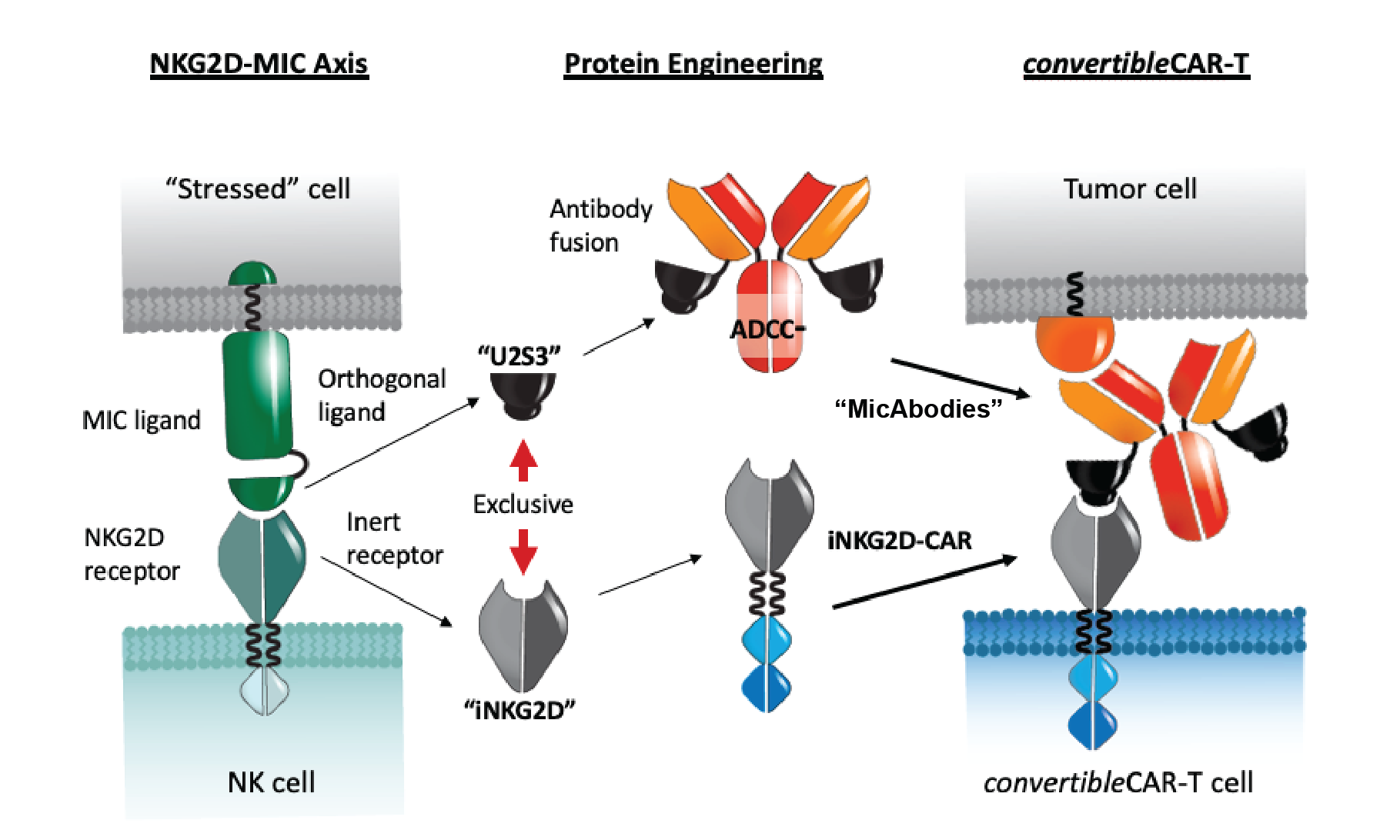
Next, we turned our attention to the MIC ligands and used a phage display-based strategy to identify variants with very high specificity and affinity for iNKG2D but not wild-type NKG2D. Essentially, we created a novel bio-orthogonal interaction. These MIC ligands can be attached to a tumor cell-targeting antibody to form a ‘MicAbody’ (Figure 2).
It is also possible to create a bivalent format, with two copies of the ligand on the bispecific molecule, while maintaining selectivity. Regardless of whether there is a heavy chain or light chain fusion on the targeting antibody we retain high binding to the iNKG2D (below picomole level) with no binding to the wild-type NKG2D.
When tumor cells are co-cultured with convertibleCAR T cells, the CAR T cells are incapable of recognizing the tumor cells until the appropriate tumor-targeting MicAbody is introduced, prompting cytokine release and other measures of activity (Figure 3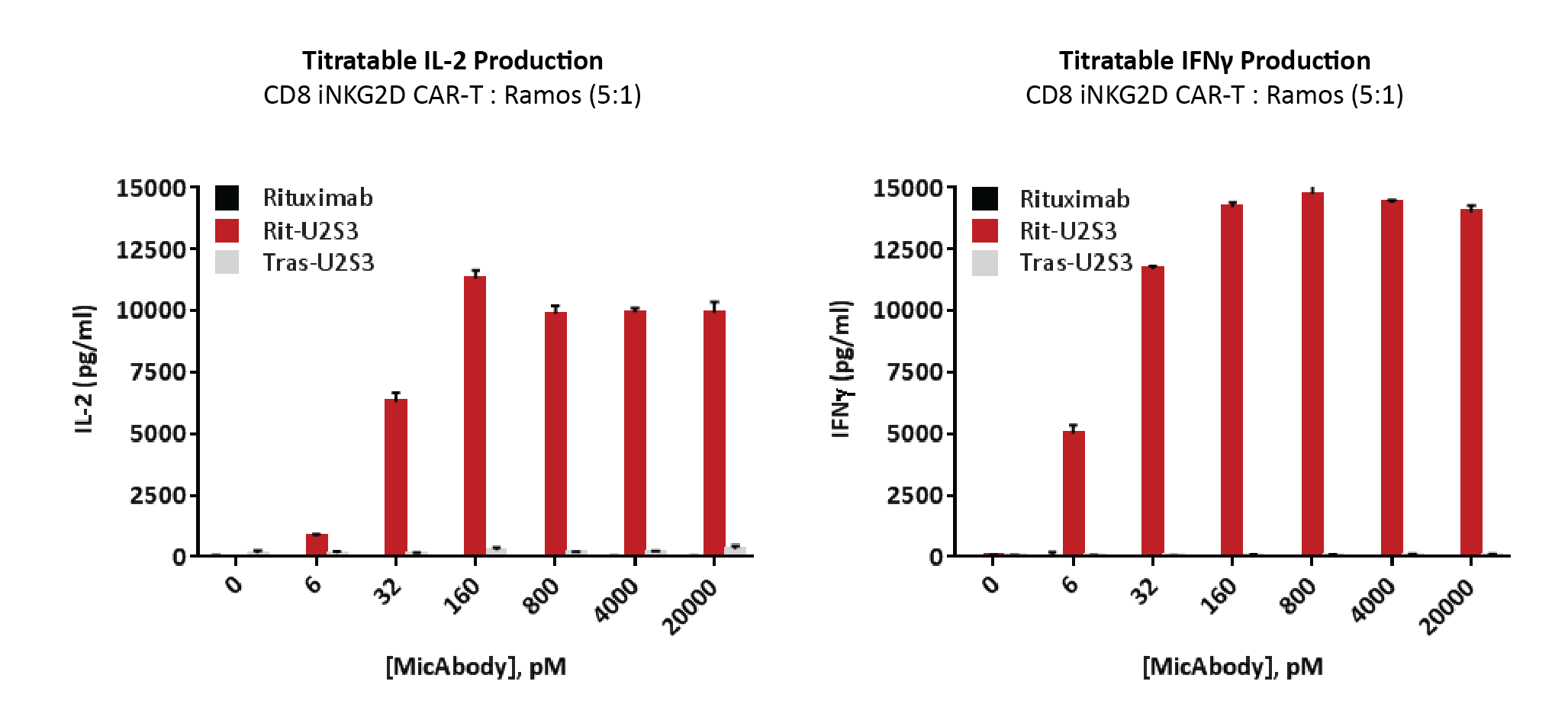
Multiplexing capabilities
Receptor saturation occurs at 5 nanomoles of MicAbody, with cytotoxicity seen at concentrations as low as 30 picomoles. That leaves a lot of space on the surface of the convertibleCAR T cells to combine multiple MicAbodies. An example can be seen in Figure 4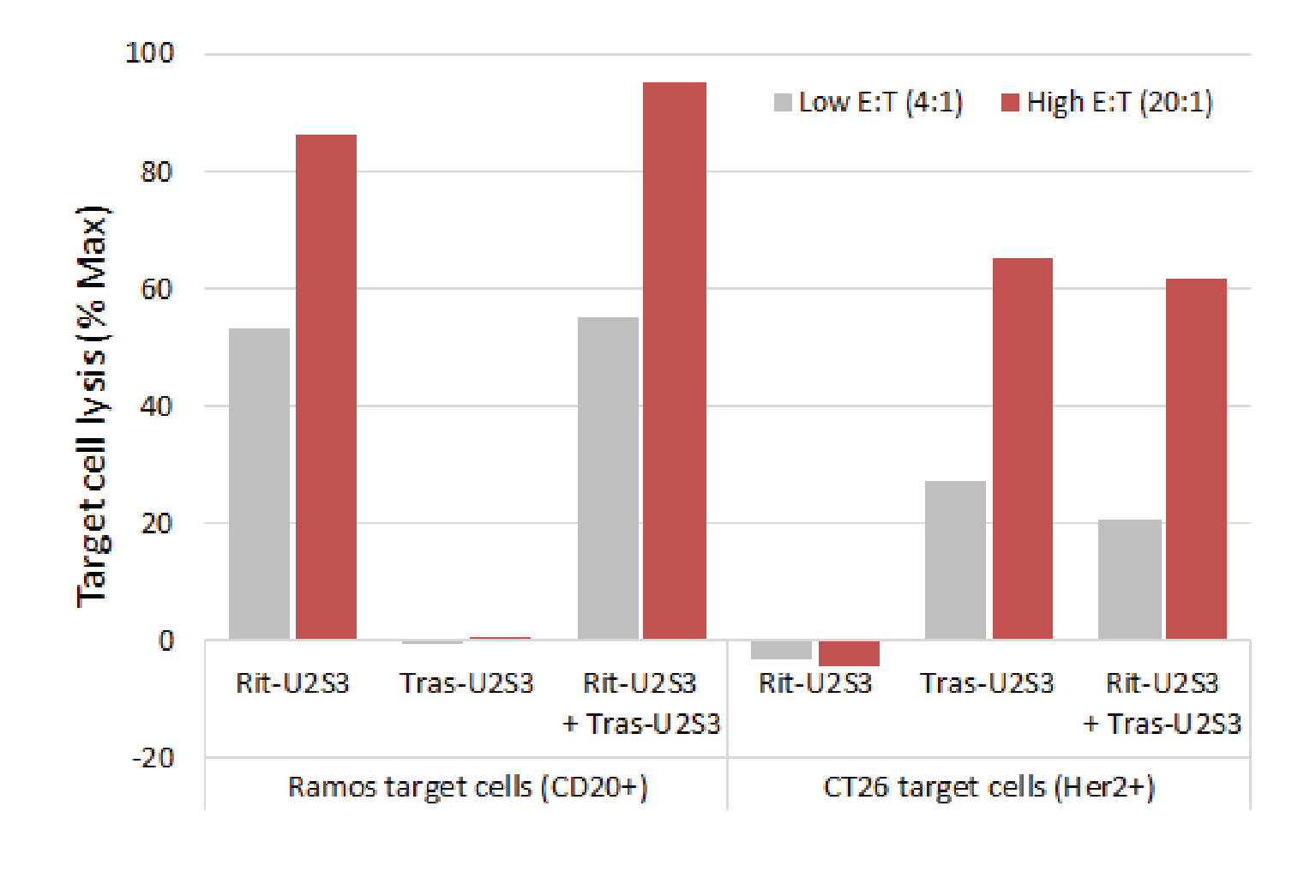
In an animal model of B cell leukemia, treatment with rituximab CD20 targeting MicAbodies plus convertibleCAR T cells was well tolerated and led to robust tumor control, and we hope to start our first clinical trial in 2022.
Manufacturing convertibleCAR T cells
Carlos Yuraszeck
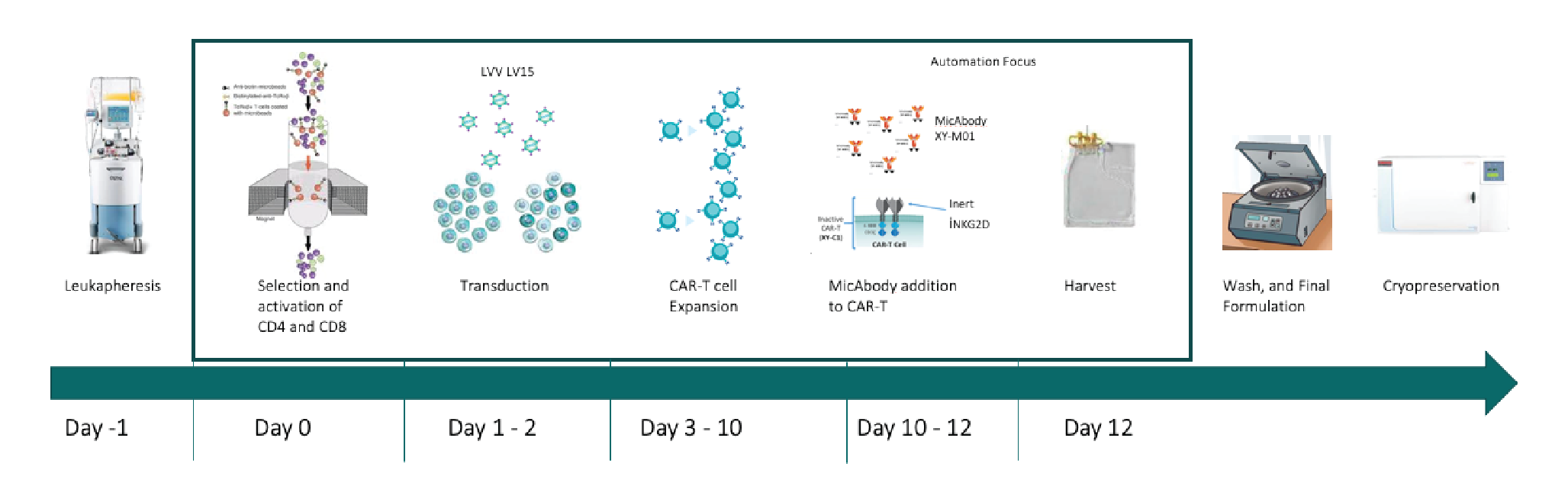
Figure 5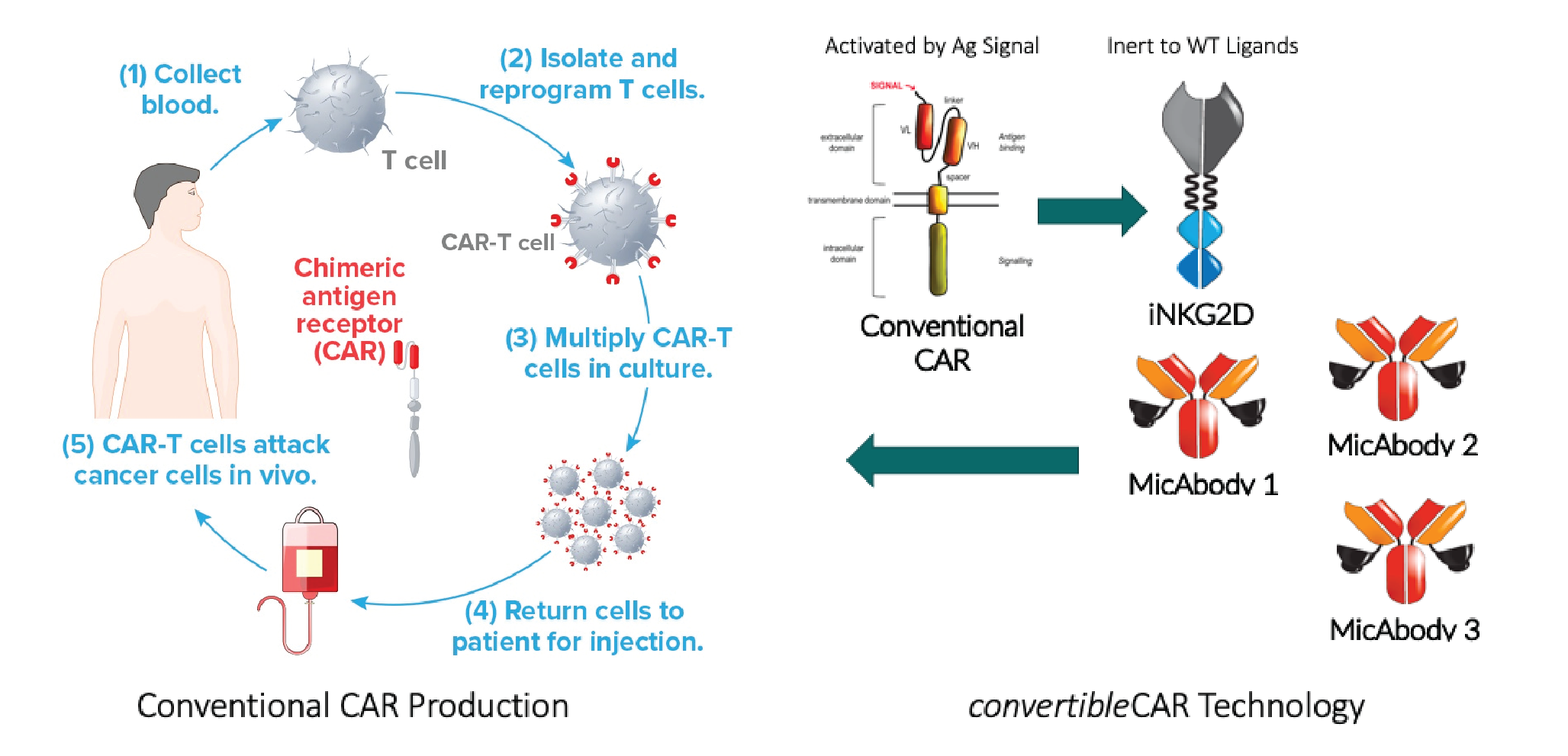
Why automate?
Compared with traditional pharmaceuticals, the clinical development time for these cell therapies is much shorter, leaving very little time for process development, or chemistry, manufacturing, and controls (CMC). That means that manufacturing decisions must be made early, process translation must be quick, and the system must be scalable from Phase 1 trials to commercial manufacturing. Time is of the essence and you don’t want to lose time changing the process between trials.
An important factor for us as cell therapy manufacturers is that moving biological materials across borders can be easily disrupted (as highlighted by the impact of the COVID-19 pandemic), making centralized manufacturing very challenging. For that reason, we believe a regional manufacturing model will serve us best. However, a major challenge of regional production is compatibility – proving that products manufactured at different sites are the same. Automation can improve confidence in this regard.
For Astellas, another consideration was that we are working on a number of different cell types, with different targets, and finding a flexible solution that can be applied across many different products would be highly advantageous for us.
Considering all these factors, we felt that automation was the right choice for us, but with a platform that also provides flexibility to accommodate our various requirements.
Having decided to automate the process, we set several criteria for selecting our preferred system. Our top five reasons were:
- Good technical support in both process and analytical development
- A closed system, to reduce the potential for contamination
- A customizable, flexible platform
- Product quality and yield within the required range (>1 × 109 CAR+ cells)
- Clinical use experience
Applying those criteria, we opted to use the Cocoon Platform from Lonza, due to its flexibility and small footprint. Lonza brought great depth and breadth of experience in manufacturing T cells and autologous products, and the collaboration has been seamless.
With a closed, automated process in place, we are confident that we will be able to deliver our convertibleCAR T cell therapies to patients around the world.
Automating the process with Cocoon
Joseph O’Connor
It quickly became clear that the biggest risk of translating the process into Cocoon Platform was the non-standard MicAbody unit operation. Therefore, this operation needed to be assessed, optimized, added to the automated protocol, and later tested.
The targets for successful process translation were:
- Subject dose greater than 800 million CAR+ cells
- Transduction efficiency greater than 50%
- Greater than 70% viability post-thaw
- Purity of over 95% CD3+ cells
- Efficient wash-out, with less than 0.1 nanomolar of MicAbody remaining post-harvest
- More than 20% of receptors occupied by the MicAbody, proving the efficacy of that unit operation
In automating the MicAbody unit operation, there were some special considerations. For one, MicAbody arming appears to be temperature sensitive, so we used cold reagents. Everything was done at room temperature and any heating elements were turned off. Plus, to speed up the process, the standard harvest protocol was modified. The automated protocol is outlined in Figure 7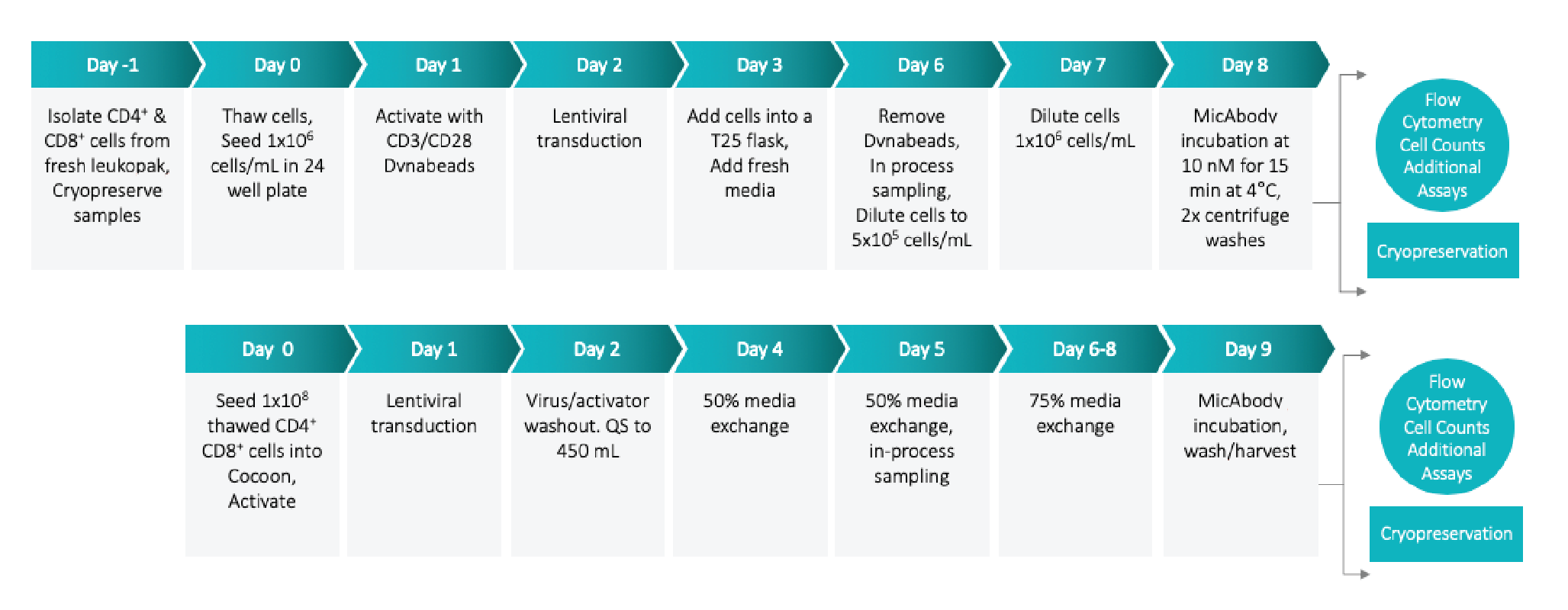
We have now performed four Cocoon runs of the Xyphos process. Cell viability was within the target range and cells from Cocoon runs exhibited increased transduction efficiency compared with manual runs (Figure 8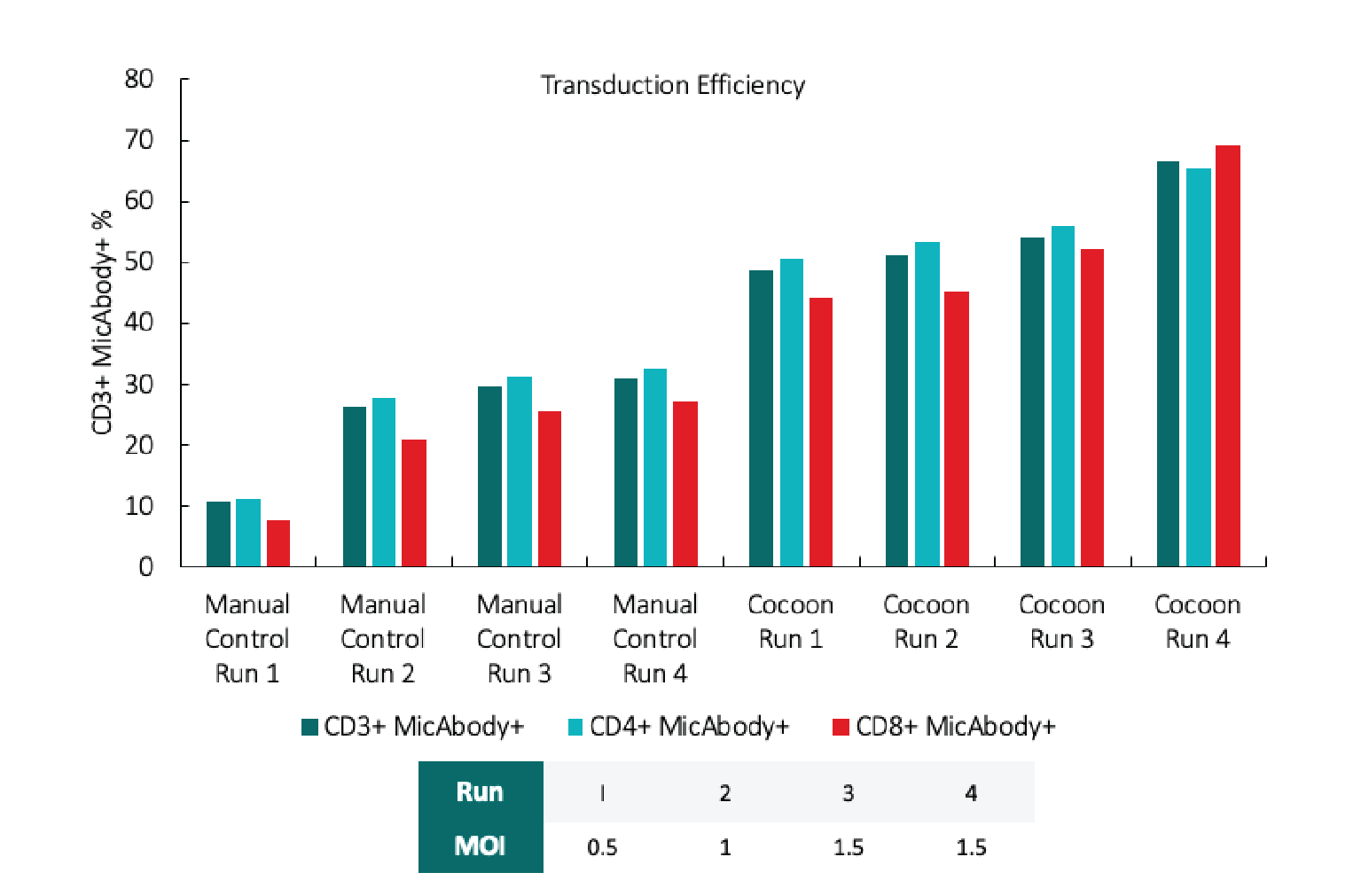
We are still investigating the reasons behind this increased transduction efficiency, but we believe it may relate to the increased surface area of the cassette growth chamber and/or the ability to mix cells periodically during transduction.
One of the most important assays is the MicAbody receptor occupancy assay (Figure 9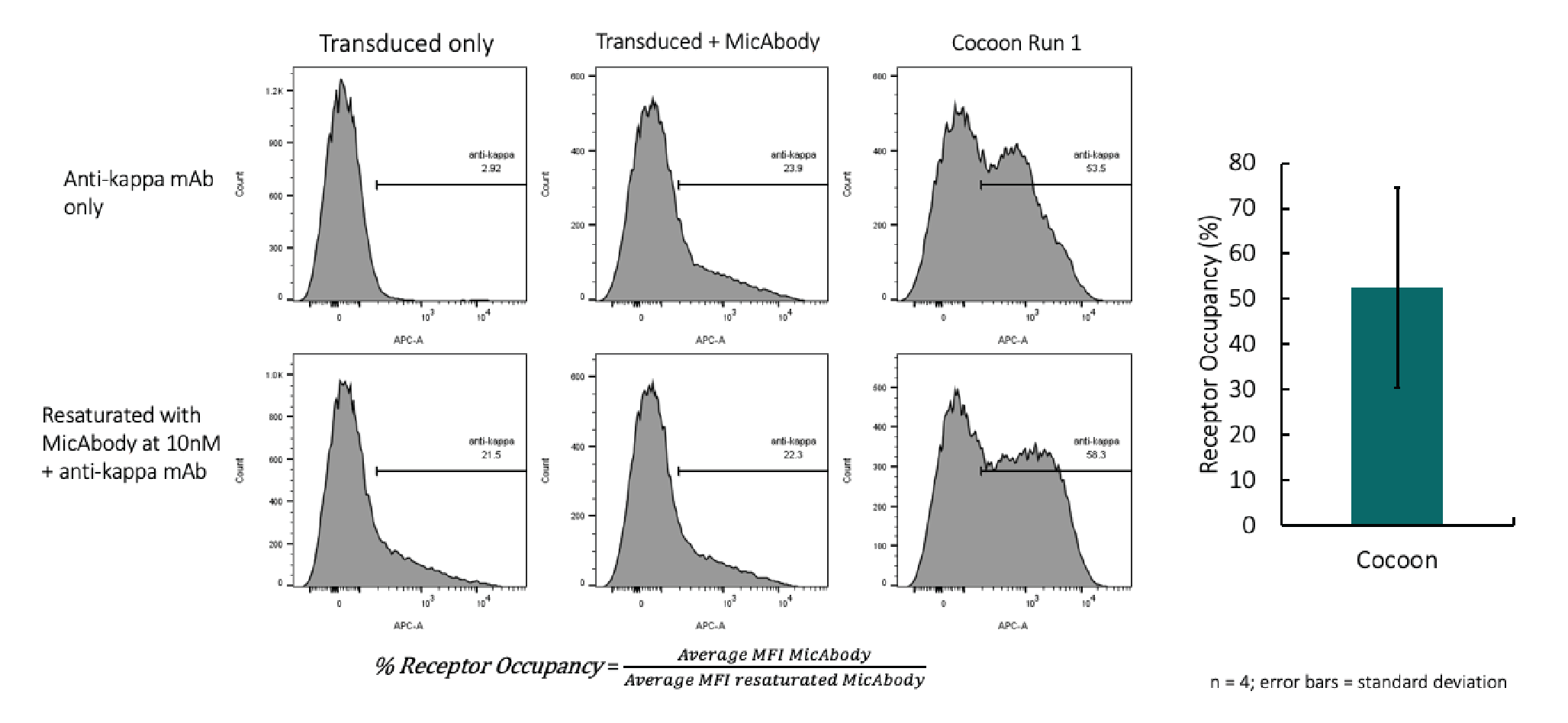
The concentration of non-bound MicAbody was about 0.015 nanomolar, which passes the acceptable criteria by almost an order of magnitude and proves we have effective washing.
Overall, the data from the four Cocoon runs (with two donors) was highly reproducible (Table 1) and met all the translation targets.
| Box 1 |
Enabling cell and gene therapies: from concept to patient.Introducing The Cocoon Platform from LonzaJoseph O’ConnorThere is wide agreement among cell and gene therapy manufacturers that automation will play an important role in the future of the field. Our survey of cell and gene therapy professionals suggests that the key considerations for implementing automated manufacturing are:
The Cocoon consists of three primary components.
Scale-outAs more autologous cell therapies are approved for larger patient groups, scaling up/out manufacturing will become a key challenge. To treat 10,000 patients per year, you would need to initiate 30 new patient processes per day on average, meaning you have 330 patient processes running in parallel. Manual production at that scale would require around 1,700 full-time employees – automation is needed to make scale-out feasible.Some companies are investing large sums to create facilities that can manufacture 4,000 patient doses annually, but our vision for autologous cell therapy manufacture involves moving to a much smaller footprint with the Cocoon Tree – an array of units on a central vertical axis, with each individual Cocoon representing a separate process (Figure 1). |
| Table 1. Cocoon platform translation data summary. | |||||
| Test | Success criteria | Cocoon Run 1 MOI = 0.5 45 IU/mL IL-2 Donor A | Cocoon Run 2 MOI = 1 45 IU/mL IL-2 Donor A | Cocoon Run 3 MOI = 1.5 100 IU/mL IL-2 Donor B | Cocoon Run 4 MOI = 1.5 100 IU/mL IL-2 Donor B |
| Subject dose (CD3+MicAbody+ cells) | >8 x 108 | 1.16 x 109 | 1.00 x 109 | 1.09 x 109 | 1.24 x 109 |
| % Transduction efficiency | ≥50% | 48.7 (control = 10.7) | 51 (control = 26.2) | 54.2 (control = 29.5) | 66.4 (control = 30.9) |
| % CD3+ cells | ≥95% | 97.5 | 97.4 | 96.5 | 98.2 |
| Concentration of non-bound MicAbody (nM) | <0.1 nM | 0.048 | 0.002 | 0.000 | 0.009 |
Q & A



Carlos Yuraszeck, Kaman Kim and Joseph O’Connor answer your questions about convertibleCAR T cells, cell manufacturing automation and the Coccoon Platform.
Q Does using a two-part system of mAbs and CAR T mean a more difficult regulatory path, because now there are two drug products, or is the final frozen cell product considered a single drug product?
CY: The strategy that we’re pursuing, which has to be yet proven, is that this is a single agent. It’s in two parts, but both the INKG2D receptor and MicAbody alone are inert.
Q What was the origin of the convertibleCAR T/MicAbody system?
KK: A lot of so-called switch or adaptor-based technologies raise concerns with regards to immunogenicity. If you start with something as human as possible, that is less likely to be an issue, so we reviewed a number of human receptor-ligand pairings, and focused particularly on those with a lot of structure-function information in the immunology space. NKG2D MIC ended being a perfect fit.
Q Can you pool different lots or batches of cells to reach a yield, and is this acceptable to the regulatory bodies?
CY: In my personal experience, although not with this particular program, it’s common to pool batches. As each lot of material meets your specifications, there’s nothing in the regulations that says they can’t be pooled and given as a single dose.
Q Can you retrieve in-process samples from the Cocoon to monitor cells?
JO: Yes, you can sample cells or sample media.
Q Are there regulatory concerns with the Cocoon Tree having multiple patients’ cells in the same space?
CY: I don’t think that’s a concern only with the Cocoon. The current standard approach is manufacturing multiple patients in the same space, so the issue goes beyond whether you are automating or not automating.
I’m a big fan of the ISBT 128 standards, which help not only with the manufacturing but also in moving products from the clinic and into manufacturing, then back to the clinic.
It’s likely that regulators will come on board to manufacture the same therapy with different patients in a closed system like the Cocoon. What we generally hear from regulators is that it’s the use of different vectors in the same suite that gives them pause. Lonza is working on experiments to prove that this shouldn’t be a concern, but ultimately, we may need to consider suite set up for manufacturing one therapy. Multiple suites for multiple therapies.
Q Is there published data using the Cocoon platform?
JO: Not yet. There are some white papers out, but we are actively working, myself included, in giving some out this year.
Q How does harvesting cells work in the Cocoon system?
JO: The cells are currently settled along the proliferation chamber so we can remove media without disturbing the cells. We add and remove buffer several times without disturbing the cells. We then use the Cocoon to rock back and forth to resuspend the cells, then collect the cells in an output bag. All the user really needs to do is press go, and then wait.

Authorship & Conflict of Interest
Contributions: All named authors take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Acknowledgements: None.
Disclosure and potential conflicts of interest: Dr Kim holds two patents: US 2019/0300594 A1 and US 2020/0138866 A1.
Funding declaration: The authors received no financial support for the research, authorship and/or publication of this article.
Article & copyright information
Copyright: Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0 which allows anyone to copy, distribute, and transmit the article provided it is properly attributed in the manner specified below. No commercial use without permission.
Attribution: Copyright © 2021 Lonza. Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0.
Article source: Invited.
Revised manuscript received: Aug 4 2021; Publication date: Aug 11 2021.
- Q & A
- Kaman Kim
- Carlos Yuraszeck
- Joseph O’Connor
- Q Does using a two-part system of mAbs and CAR T mean a more difficult regulatory path, because now there are two drug products, or is the final frozen cell product considered a single drug product?
- Q What was the origin of the convertibleCAR T/MicAbody system?
- Q Can you pool different lots or batches of cells to reach a yield, and is this acceptable to the regulatory bodies?
- Q Can you retrieve in-process samples from the Cocoon to monitor cells?
- Q Are there regulatory concerns with the Cocoon Tree having multiple patients’ cells in the same space?
- Q Is there published data using the Cocoon platform?
- Q How does harvesting cells work in the Cocoon system?