Addressing the challenges of purification and quality control in gene therapy
Cell & Gene Therapy Insights 2021; 7(6), 871–889
10.18609/cgti.2021.089
Recombinant adeno-associated virus (rAAV) vectors are a highly promising mechanism for therapeutic gene delivery. However, most industrial cell lines exhibit inefficient gene packaging, which results in a heterogeneous population of vectors composed of empty, partially-loaded, and fully-loaded capsids. Purifying, identifying and quantifying these different species is vital from a production and quality control standpoint. Improperly loaded viral capsids do not produce the desired therapeutic effect, but may still elicit an unintended immune response. This article and expert panel discussion will focus on a variety of pertinent topics in rAAV process development, with a focus on the benefits of analytical ultracentrifugation for vector purification and characterization.
Ultracentrifugation for the purification of gene therapy products
Robust and reliable purification and characterization of AAV vectors is essential to the gene therapy industry. Analytical ultracentrifugation (AUC) can offer a high-resolution purification technique, along with baseline separation between empty, full, and partially loaded capsids, and quantitation of the presence of higher-order capsid species.
Ultracentrifugation: an overview
Density gradient centrifugation (DGC) is conducted in a column of liquid medium of varying density (viscosity), and the components in the sample are separated based on their physical properties – size, mass, and density. The sample is centrifuged at a low speed of a few hundred gravity acceleration equivalent.
Two characteristics of the solution being separated are critical. First is gradient viscosity, which affects particle migration rate. The standard rule is that more viscous solutions lead to slower migrating particles. The second parameter is gradient density, which affects particle position – where the particle will finally be located vertically within the tube if spun for a long enough time.
Density gradient ultracentrifugation (DGUC) is based on the same process and relies on the same physics. The difference is the acceleration, which usually exceeds 100,000 x g. DGC is typically used to separate or characterize particles going down to ~0.1 microns in size. In contrast, DGUC can separate particles of less than 200 nanometers in size, allowing for the purification of exosomes, vectors, viruses, plasmid DNA, antibodies, and even proteins. DGUC enables consistent, high-purity separation between biologics that are very close in density.
DGUC: capabilities
Triple-layered versus double-layered viral particles have a density difference of just 0.02 g/mL and can be separated via DGUC. Looking at stable isotope labeling, even smaller density differences of 0.0036 g/mL can be separated. The ability to perform these separations is due to many viral particles having different ratios of proteins to nucleic acids. AAVs, for example, have variable nucleic acid loading, while others have variable protein shells. Proteins are generally less dense than nucleic acids, and therefore, a different ratio of nucleic acid versus protein can change overall density.
Ultracentrifugation separation methods
Figure 1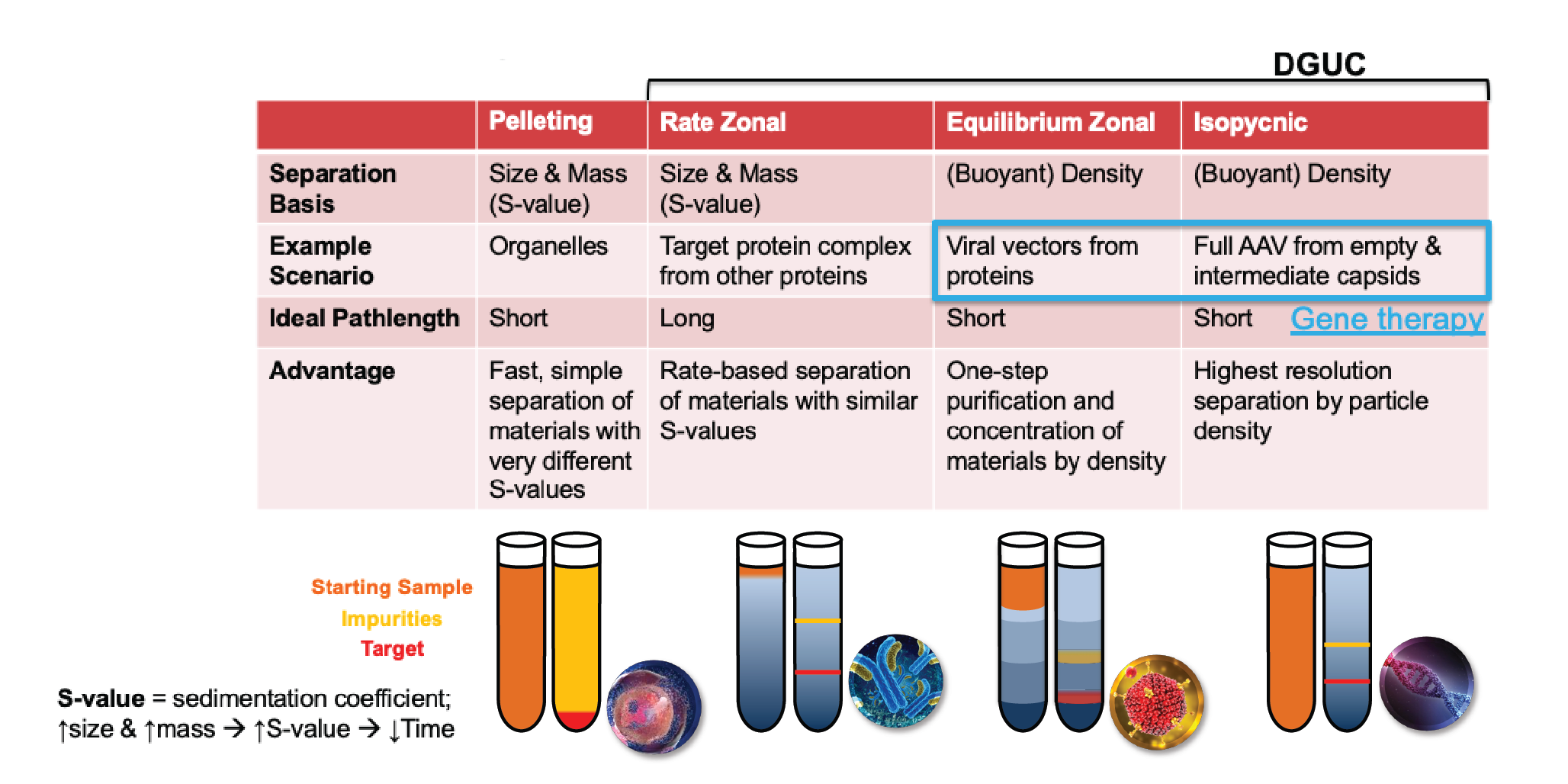
Rate zonal DGUC is an enhanced version of pelleting, where the separation basis is the same, but with the addition of a high-density material inside the tube to make the path length much longer, and therefore give better resolution.
More sophisticated than this is equilibrium zonal ultracentrifugation, which results in layers of the gradient-forming material with different densities. This is a one-step purification process and can be used to purify viral vectors.
Isopycnic, or buoyant density separation, utilizes an infinite number of density steps. This is also an equilibrium technique and gives the highest resolution it is possible to have in a density gradient experiment. Both equilibrium zonal and isopycnic DGUC are used for gene therapy, but isopycnic provides the highest resolution.
Case study: AAV purification using iodixanol versus cesium chloride gradient
When selecting a material to form gradients inside a tube for DGUC, iodixanol and cesium chloride (CsCl) are two common choices – and both offer certain advantages (Figure 2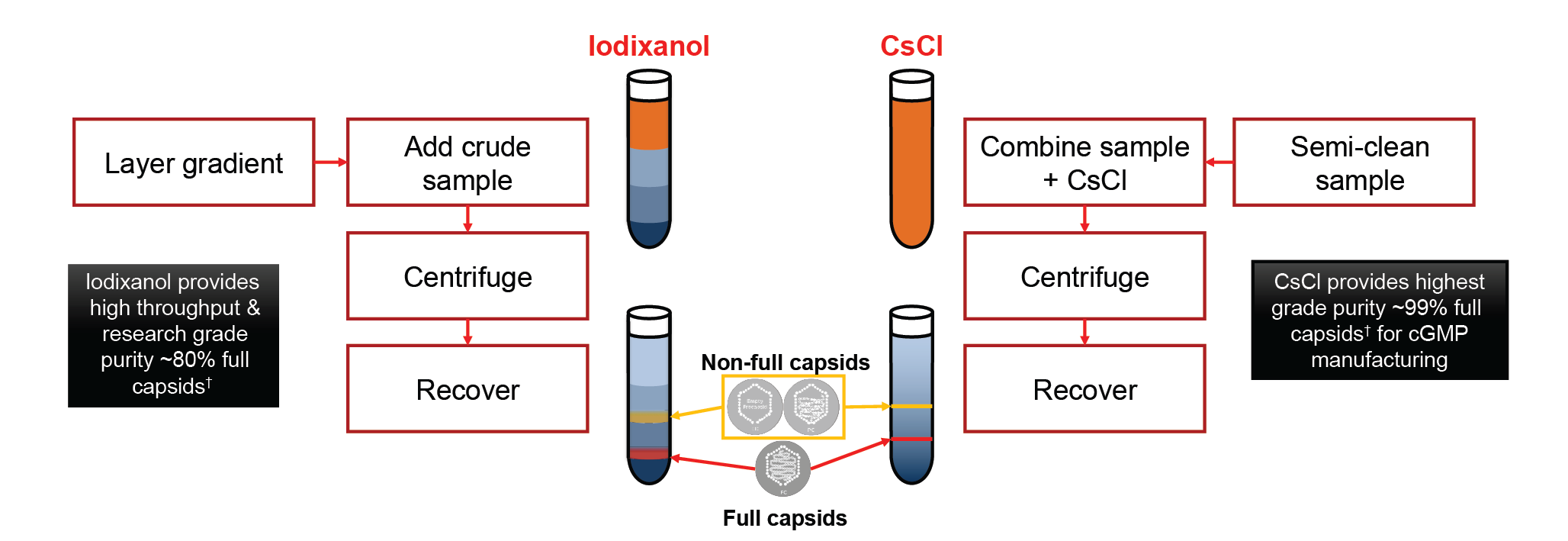
Iodixanol provides a relatively shorter spin time and therefore gives higher throughput. It can provide research-grade purity material, typically with up to around 80% full capsids [1].
Cesium chloride requires a longer spin time but will result in the highest possible purity, suitable for cGMP manufacturing processes. Using this material it is possible to achieve 99% full capsids [1]. The other advantage is that you can load significantly more material in a cesium chloride experiment.
AAV characterization via AUC
The key quality control questions in AAV production include:
- What percentage of viral capsids are intact, and how many have broken down?
- Can intact but empty viral capsids be distinguished from intact viral capsids that contain the target genetic material?
- Can the presence of partially loaded viral capsids be quantitated?
Analyzing samples via analytical ultracentrifugation (AUC) can provide answers to these questions. If you consider a snapshot halfway through the experiment, you can see that the sample sector shows three distinct regions, as shown in Figure 3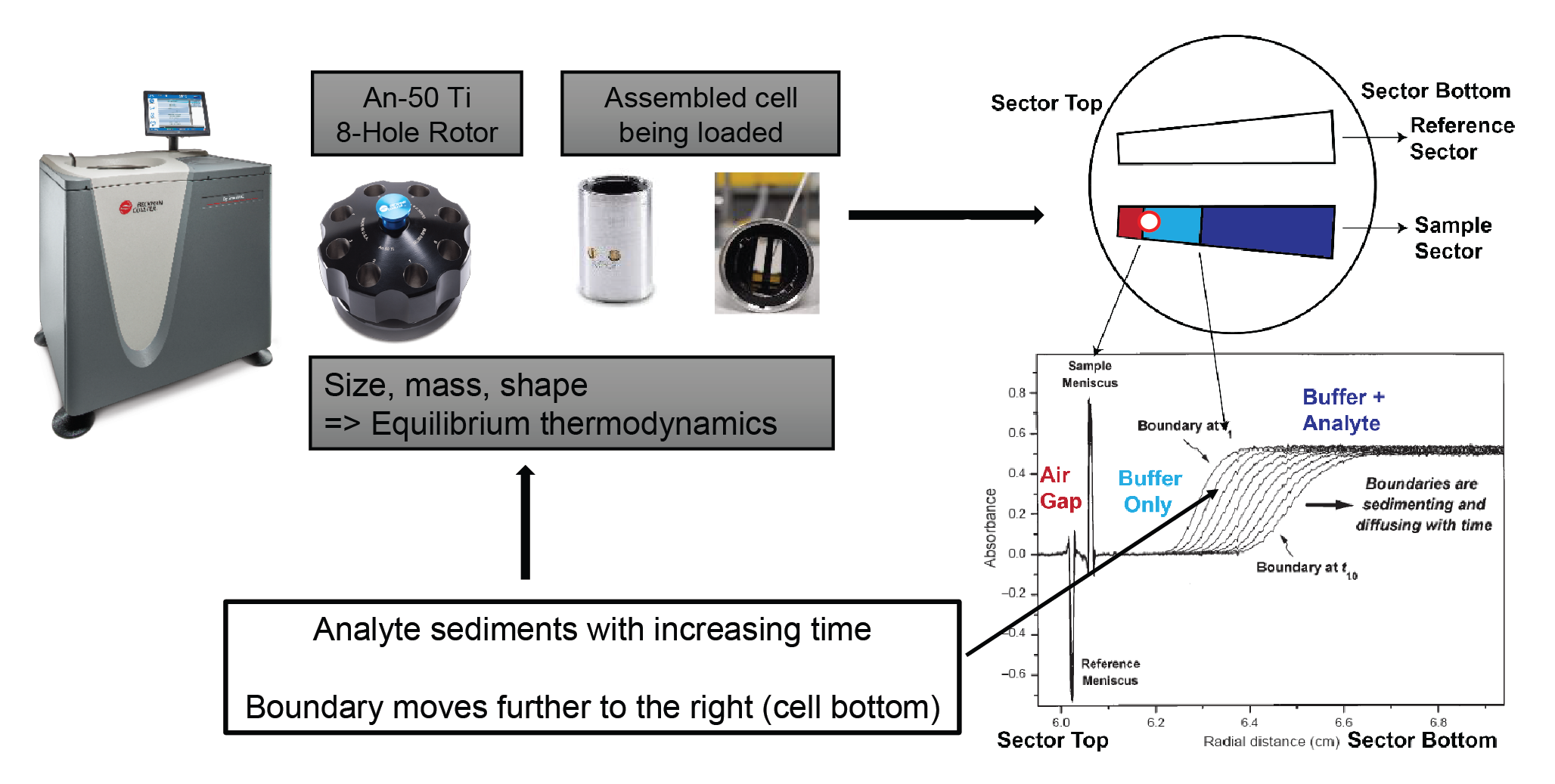
As an experiment progresses, the boundary moves further down, towards the bottom of the cell (Figure 4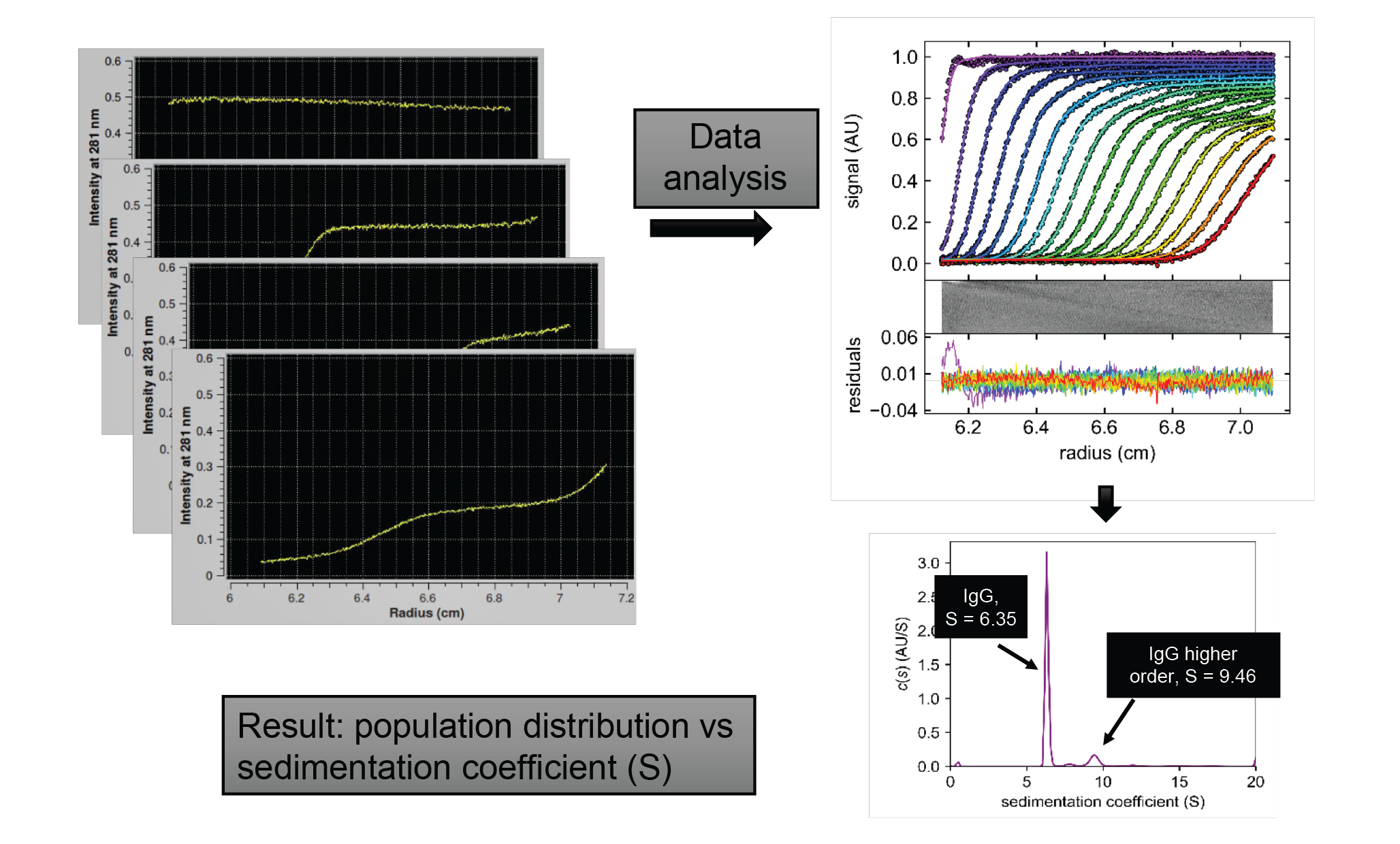
AUC in viral vector quality control
Figure 5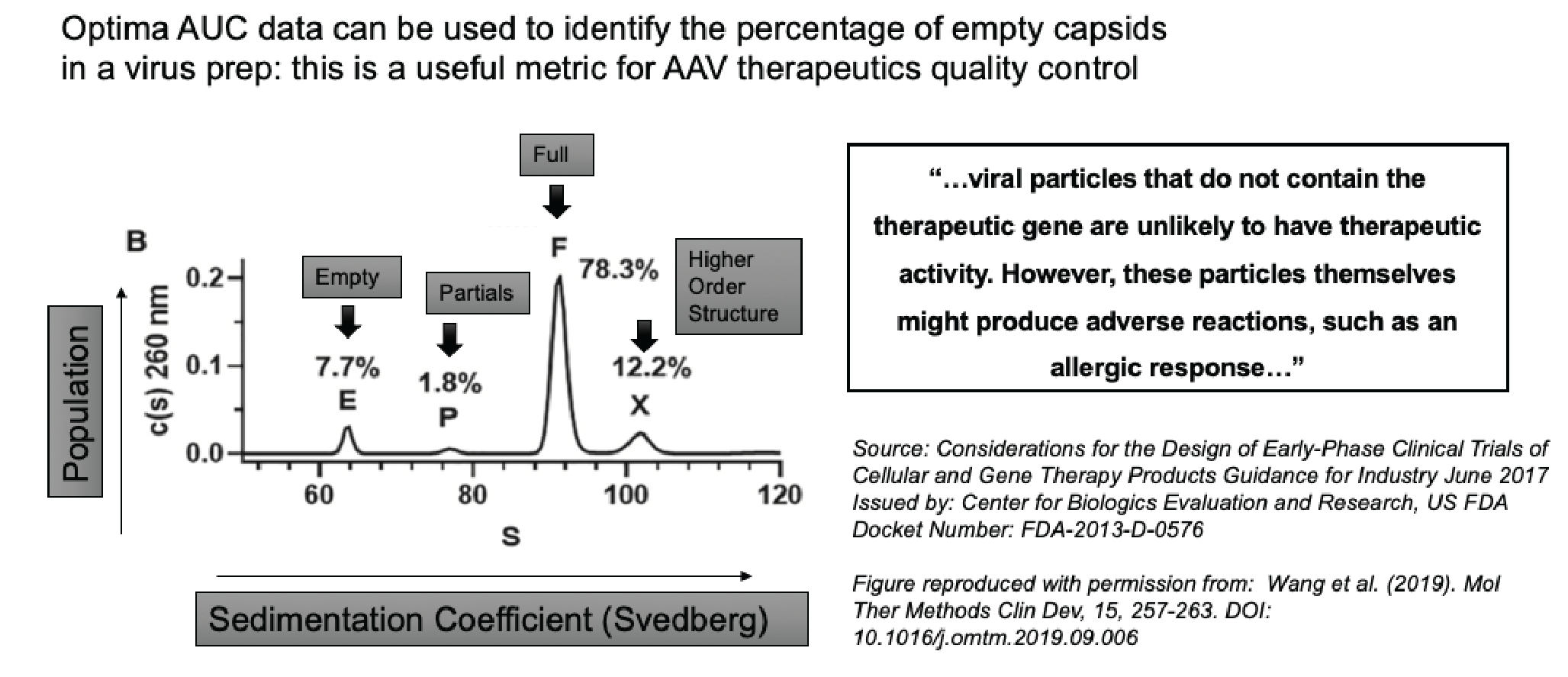
It is vitally important to distinguish between all of these different species because as FDA guidance points out, viral particles which do not contain the therapeutic gene are unlikely to have a therapeutic effect. However, the particles themselves might produce an adverse allergic response.
Vigene reference studies
The following results come from experiments that were performed on a library of AAV reference standards produced by Vigene Biosciences. The raw data and analysis can be seen in Figure 6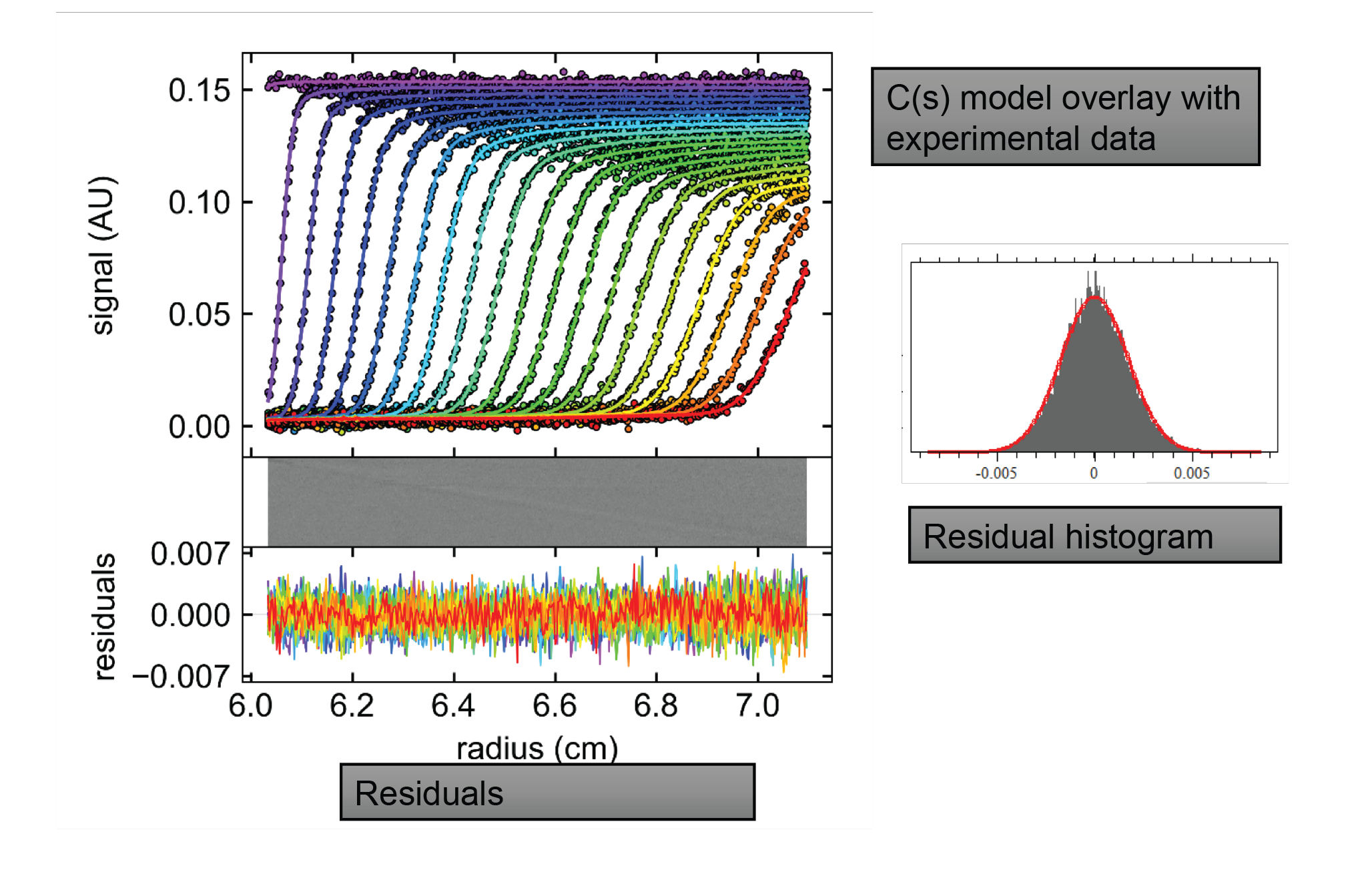
A population distribution is shown in Figure 7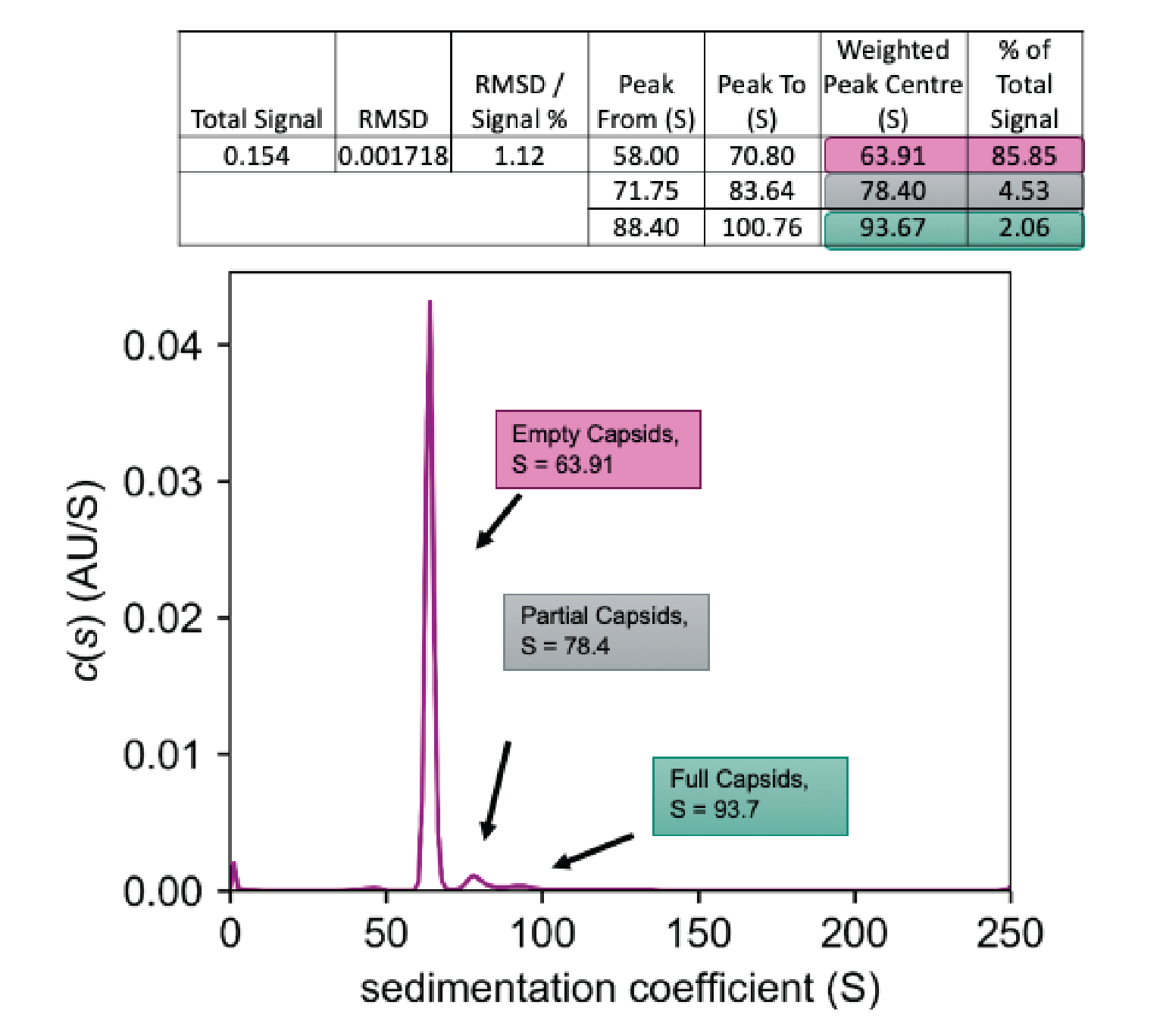
Figure 8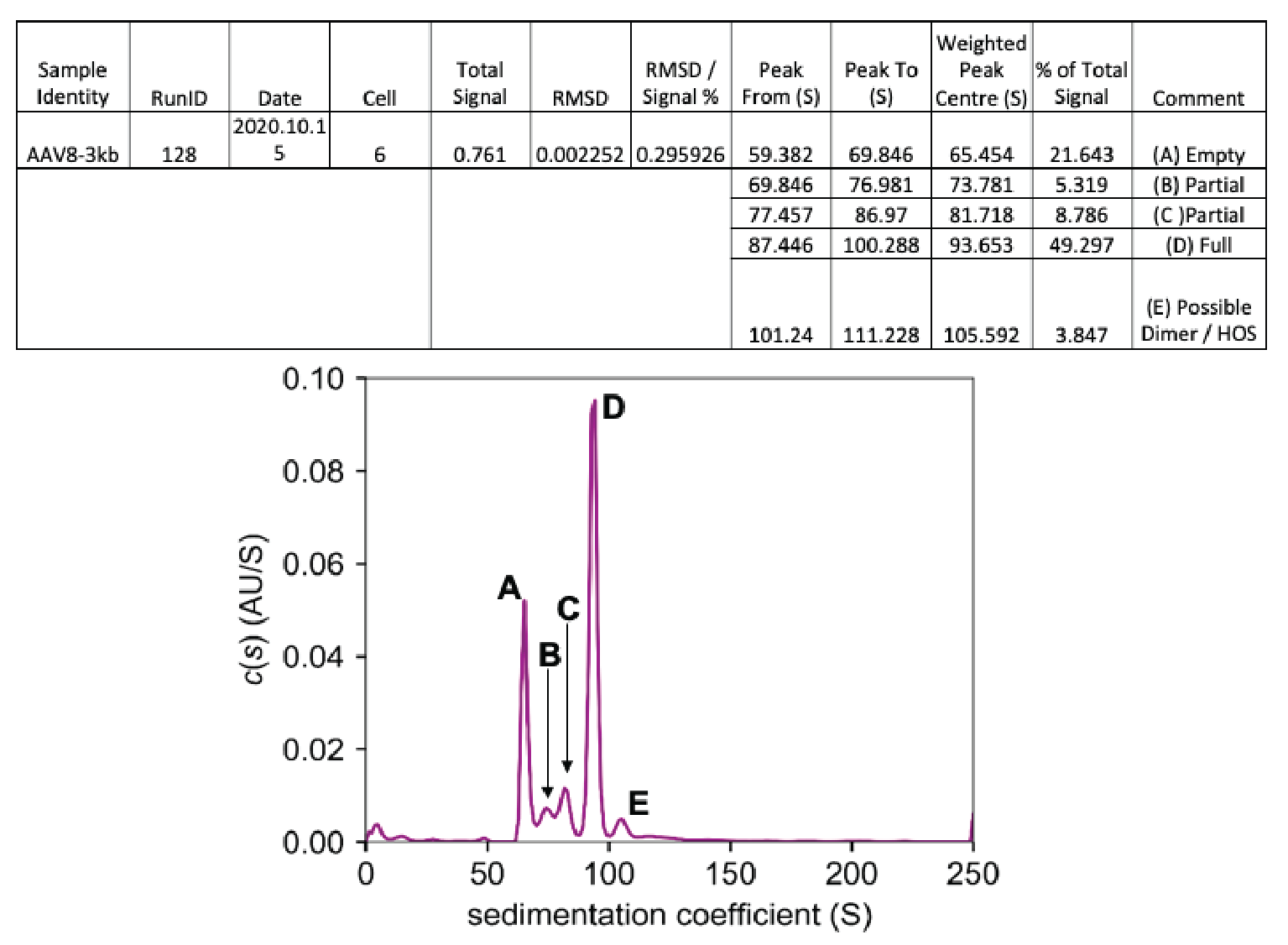
Again, a number of different species are seen, all of which are labeled. Empty capsids show up at around 65.5 Svedbergs, making up almost 22% of the total signal. The full capsids show up as peak D at 93.65 Svedbergs, making up slightly over 42% of the total signal. In this example, we have not one but two partially loaded species, which can be identified via an AUC experiment, shown by the peaks marked as B and C. These contain 5.3 and almost 8.8% of the total signal, respectively. Finally, at 105.6 Svedbergs some kind of higher-order species of capsid is seen.
To summarize, AUC is capable of providing baseline separation between different capsid species. It is capable of identifying not just partially loaded capsids, but also higher-order species, and of distinguishing between and quantifying the relative percentage of empty and full capsids.
Insight
DGUC is a high-resolution purification technique that can be utilized for critical biotherapeutics. Cesium chloride DGUC can provide cGMP-grade product when purifying AAV, and can provide up to 99% full AAV capsids. In contrast, iodixanol DGUC can provide research-grade material, with up to 80% full AAV capsids, and slightly higher throughput.
AUC performs a native-state solution-phase analysis to quantitate the different species of AAV capsids in a mixture, and can also provide a capsid loading fraction that represents the empty/full capsid ratio. AUC experiments can provide baseline separation between empty, full, and partially loaded capsids, and can also quantitate the presence of higher-order capsid species.
Finally, AUC experiments are processed and analyzed using the same experimental and data analysis protocol, completely independent of knowledge of both the genotype and the gene of interest. This means that an AUC experiment and analysis protocol that has been optimized for AAV2 can also be used with AAV5 or AAV7, with no need to redesign the experiment or protocol.
Reference
- Strobel B, Miller FD, Rist W, Lamla T. Comparative Analysis of Cesium Chloride- and Iodixanol-Based Purification of Recombinant Adeno-Associated Viral Vectors for Preclinical Applications. Hum. Gene Ther. Methods 2015; 26(4): 147–57. Crossref
- Wang C, Raju Mulagapati SH, Zhongying Chen Z et al. Developing an Anion Exchange Chromatography Assay for Determining Empty and Full Capsid Contents in AAV6.2. Mol. Ther. Methods Clin. Dev. 2019; 15: 257–63. Crossref
Q&A




Q (SS): Our discussion will be primarily focused on the production and quality control of gene therapies, but we have also received some questions on Akash’s presentation. Have these partially filled capsids being analyzed by any other biophysical or structural techniques? Do we know what is inside of the capsid, and are they intact?
AB: These partially filled capsids appear to be intact. The identification and characterization of partially filled capsids remains one of the big mysteries in the field at this current moment.
What we do know, based upon some amount of commonality with orthogonal techniques such as electron microscopy, is that the partials do appear to be intact capsid species. However, the identity of the genomic content inside the partial is something that still remains to be uncovered.
Q (SS): Moving on to a relatively broad question – what do our panelists view as the biggest challenge facing the field of gene therapy today?
AB: The biggest challenge in the field of gene therapy today is the question of cost. Preparing gene therapy products is very expensive because of the workflow, the number of different steps involved, and the sophistication of the product that you are making, which requires equally sophisticated characterization. Anything that the biotech field can do to bring the expense down is ultimately the direction that we want to go in.
AC: It is not a well-characterized process, and that is probably one of the biggest contributors to the costs associated with it.
As we move forward as an industry, we need to have a well-characterized process, following a full understanding of each of the manufacturing steps. This is where the QC testing of in-process characterization becomes very important as well.
LK: Looking at this from a slightly different angle as a reagent provider, I would say one of the largest problems is production capacity. Process improvements need to be made because we are not currently able to meet the growing demands in terms of the desired speed to the clinic and the sheer volumes needed per patient dose.
Part of that is due to insufficient resources like plastics, media, GMP-grade plasmids, and high-performance reagents. Essentially, everyone is fighting for those same limited resources.
Of course, there is also a significant challenge in understanding patient dose requirements, with the lack of reliable in vitro and in vivo validation methods.
KR: From our perspective, the main challenge is still to generate a stable product suitable for commercial use. After that, the consistency of the production is also not easy to achieve. The workflow is not fully established, and I think from what we are seeing, differences between samples we receive are larger compared to traditional biochemistry where we characterize antibodies. Troubleshooting when deviations are found is difficult. There are also challenges in the consistency of batches, at least in the early phases.
Q (SS): With respect to purification, Akash, what are the bottlenecks that you see, and what are some of the potential solutions for dealing with those bottlenecks?
AB: Assuming that you have either been making your viral particles yourself, or you went to Vigene with your gene of interest and used their services to make the virus particle, inevitably you run into the bottleneck of workflow and throughput.
The general steps to purify your product, with the understanding that we are mostly talking about AAV, are usually a combination of filtration, chromatography, and then ultracentrifugation.
Some of these techniques do lend themselves to relatively easy scale-up – such as the filtration and chromatography steps. On the other hand, as I discussed above, some of the very best purification that can be obtained, to give you the best possible loading fraction, comes from DGUC. This is less of a scale-up and more of a scale-out technique.
When we think in terms of scale-out, the natural question that arises is how can we improve the workflow? There are definitely opportunities to apply automation to certain steps in this workflow and make things go faster. If you do have automation coming in, then you will want to have real-time monitoring of CQAs as far as possible. Therefore, there is a fair amount of engineering waiting to happen, which I believe can transform output in this field.
Q (SS): In addition to purification, one of the primary challenges in gene therapy production is the overall high manufacturing cost. What are the primary reasons for this high cost and how can we start to bring them down?
LK: Currently, production costs for a 2,000-liter bioreactor can run upwards of 1 to 2 million dollars in a GMP setting; which is obviously very expensive. Raw materials are expensive, as is GMP suite time analysis. In addition, there just isn’t enough of any of that to support all the work that needs to be done.
I don’t anticipate the costs of media, cells, DNA, and other raw materials to decrease all that much. In our view, cost savings are attainable with process improvements that lead to higher functional virus production and higher percent full capsids, produced without extensive purification required.
This is Mirus’ goal in developing TransIT-VirusGEN® reagent formulation. While we can’t change costs of everything involved in gene therapy production, the hope is to decrease the number of runs required to meet therapeutic requirements through process development, rather than having to run three or four 2,000 liter reactors. If you can drop that down to one, it will save a significant amount of time, resources, and money.
Q (SS): Where do you think the biggest value lies in terms of optimizing viral vector production? In other words, which steps in the workflow do you think have the most room for improvement?
AC: Probably the yield – there is some really fantastic work going on with producer cell lines that get you away from the triple transfection process. There is also fantastic work going on around purification; however, you are going to have to be able to calibrate and understand these new technologies, and that is where standards come into play.
If you have a well-characterized standard, you can assess new technologies because you know what you are looking for. This is a big area, and as we move and we innovate, we have to know where we are.
Q (SS): Production is just one part of the story, and characterization is the other. Akash, can you give us an overview of some of the most popular characterization techniques that are being employed in the gene therapy field?
AB: I really like what Audrey said – as we innovate, you have to know where you are. Characterization is all about knowing exactly where you are.
We can split up the characterization tools into those which deal with the genetic payload, and those that deal with the vector or the carrier.
The tools that characterize the genetic payload are usually variants of highly evolved PCR-type techniques. I would say that digital droplet PCR (ddPCR) is the current state of the art in terms of low sample consumption and obtaining good statistics for your result.
For vector or delivery vehicle characterization, you have more or less the entire bag of tricks from biophysics available. On the low-tech end of things, the standard SDS-PAGE and ELISAs can tell you what kind of proteins you have in the capsid prep.
You could then go and do some 260/280 spectrophotometry, this is going to give you clues as to the relative amount of DNA and protein in the mixture that you have. Next, you need to understand a little bit about the particles themselves. You probably want to do some light scattering experiments to give you information about capsid size.
Next, you would probably want to look at chromatography. Analytical ion exchange chromatography is a variant of the popular separation chromatography, with different analytical inputs, and is quite user-friendly in terms of characterization and quality control.
Similarly, you could also end up doing capillary isoelectric focusing (cIEF). If you want to image the capsid, the best (and pretty much only) way to do it is with electron microscopy.
Finally, my own specialization is AUC, which looks at particle sizing but also looks at density, and therefore gives you one of the best possible ways to characterize and quantitate the binding fractions of viral capsids.
Q (SS): Leisha, can you comment on how characterization and analytics are involved in process development? Specifically, how far upstream can you take some of these techniques?
LK: At Mirus, with our customers in upstream process development, we are always looking at how different parameters impact functional virus production levels for cell types, media, plasmid design, and transfection optimization parameters – reagent to DNA ratio and so on. Then we want to analyze how this alteration in the process changes the output.
Typically, the virus at this stage is characterized by using ddPCR or qPCR to determine genome content, as well as ELISA to measure capsids. Of course, neither of those methods tells the whole story. These assays are performed regularly because they can be done quickly, on crude virus preps, which makes it convenient. But neither method is great for measuring the true potency of the virus.
This is where a functional assay comes in, where the virus is used to transduce gene expression in a relevant cell type. These are helpful but time-consuming and tedious, so we don’t typically see a lot of groups using these methods.
We are starting to see more AUC used in upstream process development. It offers a clearer picture of virus quality and quantity. What I love about it is that it is serotype-independent. Functional assays can be such a challenge because every serotype prefers a different cell type for transduction levels, and that is just messy, so AUC is really powerful there.
I would say the issue for widespread adaptation is throughput, and I am sure that Beckman is looking to address those issues. We at Mirus are certainly hoping to see that.
Q (SS): Klaus, can you describe some of the major differences between in-line analytics during production compared to end-point analytics for the finished product?
KR: There are wildly different approaches that have to be used in these two aspects. The samples that come from in-line analytics or from optimizing a production process are of non-standard quality with respect to concentration and purity. When the analysis needs to be done, we take what we get, and we have to deliver the result fairly fast. The client doesn’t want to wait very long at that step, and wants to see the result of the process, so that needs to be delivered.
Also, the sample concentration is not in the optimal range, and there are typically more impurities present in these samples. AUC must – and can – deal with all of this. This is a good thing about in-line analytics samples: they are a little bit of a surprise every time.
When we characterize the final product, this is of course done based on a method description, and we follow that strictly. The product has a defined quality, and surprises don’t happen very often. We know what we can expect from the sample.
So there are two very different approaches we can take in these situations – but AUC can do that, and other methods could be applied to guide that.
Q (SS): Akash – how do you qualify and validate an AUC method?
AB: The answer to qualification and validation is statistics, statistics, and then some more statistics.
If you are running an AAV sample, you don’t have to worry about the phenotype dependence of the experiment too much because, as Leisha pointed out, AUC is a serotype-independent technique.
However, you do want goalposts. You want to know what an empty capsid looks like and what a full capsid looks like. If you have your own internal reference standards and you run experiments on that at the speed that you want, you have statistics and you are good to go.
If you don’t have your own internal reference standards, then you use Vigene’s reference standards. You can also do this in order to define the goalposts of what empty and full capsids look like.
Once you have done that, you are basically in business. The method is validated and you are ready to run lots of these experiments, with maybe one reference sample in each run to get that validation result for every single experiment.
Q (SS): What about higher-order species and aggregates?
AB: You would quantify higher-order solutions and aggregates just like you would quantify anything else in an AUC experiment, because the number of the AUC output lends itself to direct integration. This again uses your reference standards in order to define the goalposts. If you have done your orthogonal experiments, you know that the genome-loaded capsid sediments within a specific range of sedimentation values. When you start seeing solutions turning up at higher values, you know this is higher-order structures – you know this is more than just your active, filled capsid.
Quantifying it is as simple as doing the integration around your species going to 105 to 110, using the same method you use for quantifying empty versus full.
To go into more detail and identify the actual content of the higher-order species, that is something that multiwavelength AUC can give you a lot of insight into.
Q (SS): Leisha, can functional/transduction assays be done on AAVs obtained from crude culture, or does the sample need to be purified? If so, to what extent does the AAV need to be purified?
LK: We routinely perform our functional QC assay using crude virus. One caveat is that we are often working with AAV2, which transduces many different cell types very well. We do a substantial dilution – 1:2,000 to 1:5,000 of that virus – which works beautifully to transduce cells.
If you are working with a serotype like AAV5, which is not nearly as good as at transducing many different cell types, you need a more concentrated sample of your virus to do the transduction. Then you may run up against the issue of having some of the components of your chemical lysis buffer start to impact the cell health of the cell type you are trying to transduce. In those cases, you may see better and cleaner results from using a batch-purified AAV sample rather than using the crude sample.
Ultimately, it depends on how well-paired your AAV serotype is with the cell type that you are trying to transduce, as well as other factors like what your AAV is specifically expressing.
It is a bit of a yes and no answer. It can definitely be done, but it is important that if you are transducing cells with something like AAV5 and want to use crude virus, you have the appropriate cell type for that transduction.
Q (SS): Audrey, can you comment on the regulatory landscape, specifically in the context of the cGMP characterization?
AC: Unlike the mAb world, we don’t have a set playbook where everyone knows the guidance document that was put out last year. Those are things we are trying to meet, but sometimes it is hard in the early phases.
The regulatory agencies do acknowledge that some of that information is limited during the early phases of development, and for us to set specifications or release testing you might have to do that at later phases as you gain more information and move forward in your manufacturing process. I do expect that bar will be raised higher as you move down the clinical path, or as the indications become more broad.
We are going to have to come up with a set of best practices. One of the best ways to do that is to come together as an industry and work towards it.
Having discussions like this, or consortiums where we come together and share best practices, will benefit all of us. The regulators will like that too, because they can then also input on these types of processes, and we will move forward as an industry.
Q (SS): Klaus – can you comment on the state of the art for cGMP characterization that you use, and what you would like to see?
KR: For some of the methods there is just not a valid GMP strategy that is a full GMP approach. In these cases, there needs to be a pragmatic approach.
Of course, for other methods there is a full GMP strategy, like high-performance liquid chromatography (HPLC) methods, micro-flow imaging (MFI), light obscuration; these are the ones we also have in-house. However, for sedimentation velocity AUC (SV AUC), there is no out-of-the-box GMP solution available yet.
There are approaches to get as close to a full GMP solution as possible, and of course this is something that we as an AUC community would like to see, but it’s just not there yet. The instrument needs to be qualified, plus the software, the audit trail… everything needs to be there.
We can work to get as close as possible and as close as the regulatory agencies want. I think the agencies acknowledge that there is a lot of effort but not a full solution yet, in several of the methods we apply.
Q (SS): Can each of you give me a summary on what you think the future of gene therapy looks like?
AB: The future of gene therapy is very bright. This technology is transformative. Even keeping in mind that these are still early days, and keeping in mind all of the caveats and warnings, there is a lot of promise.
In terms of manufacturing, that promise really comes from automation and a lot of engineering development. With that, one thing that we really didn’t speak about yet is software.
It is eventually going to be possible to start monitoring CQAs in real time. You may even get to the point where end-point assays are just a confirmation of what you already know, because you have been monitoring CQAs so much so in real time that you have been able to trace the health of a single batch from start all the way to fill and finish. You will probably also have some degree of predictive analytics, and all of this is going to bring down costs significantly.
With automation, better software, and enormous efforts from our friends in cell biology and virology to create better vectors and custom vectors, I think the future is really bright and exciting.
AC: I would echo Akash – it is a bright future. We are sort of where monoclonal antibodies were 20 years ago, when they were the new kid in town. They are now considered sort of plug and play, and by learning from the past, we should be able to get to that plug and play status a lot faster.
LK: The field will continue to focus on addressing manufacturing and safety challenges. Thinking further ahead, and assuming continued success in the treatment of additional diseases, the number and types of clinical indications addressed by cell and gene therapies will only grow, as will further development of novel capsids that increase efficacy and lower required dosages to improve safety.
I expect we will see an even greater push earlier in the development process for higher quality raw materials, which was largely the driver for Mirus to develop our VirusGen products in GMP grade. I hope that we see AUC much more broadly used earlier in the process as well, given the clear benefits that Akash outlined.
Ultimately, the hope for cell and gene therapy is to transform lives, and an even greater number of cures for previously untouchable diseases.
KR: I also see a very bright future. It is exciting to see how these platform technologies are getting developed, and how we are moving away from proteins into many different fields like liposomes, nucleic acids, and diverse viral vectors. From the analytics perspective this is also very exciting, because these require different approaches.
I see AUC playing a big role in that, since it has a very clear advantage in that it doesn’t require a matrix to achieve a separation.
Many of these viruses are very big. They are almost particle-sized, in the nanometer range. Many of the other methods just fall apart based on the functional principle, but AUC without a column matrix can still separate and characterize these samples, and help in formulation development and lyophilization product characterization, and all these processes that we work in.
Authorship & Conflict of Interest
Contributions: All named authors take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Acknowledgements: None.
Disclosure and potential conflicts of interest: Leisha Kopp is an employee of Mirus Bio LLC, a biotech company that develops and sells reagents for gene and cell therapy manufacturing.
Funding declaration: The authors received no financial support for the research, authorship and/or publication of this article.
Regulatory/Trademark Statements: All product and services identified, unless noted as for in vitro diagnostic (IVD) use, are for research use and not intended or validated for use in the diagnosis of disease or other conditions.
©2021 Beckman Coulter, the stylized logo, and the Beckman Coulter product and services marks mentioned herein are trademarks of Beckman Coulter, Inc. in the United States and other countries.
All other trademarks are the property of their respective owners.
21.08.2328.CENT
Article & copyright information
Copyright: Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0 which allows anyone to copy, distribute, and transmit the article provided it is properly attributed in the manner specified below. No commercial use without permission.
Attribution: Copyright © 2021 Beckman Coulter Life Sciences. Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0.
Article source: This article is a transcript of a previously published webinar, which can be found here.
Webinar published: Jun 24 2021; Publication date: Aug 24 2021.
This article is part of the Vectors channel, Vector characterization & analytics 2021 edition
- Q&A
- Akash Bhattacharya
- Audrey Chang
- Leisha Kopp
- Klaus Richter
- Shawn Sternisha
- Q (SS): Our discussion will be primarily focused on the production and quality control of gene therapies, but we have also received some questions on Akash’s presentation. Have these partially filled capsids being analyzed by any other biophysical or structural techniques? Do we know what is inside of the capsid, and are they intact?
- Q (SS): Moving on to a relatively broad question – what do our panelists view as the biggest challenge facing the field of gene therapy today?
- Q (SS): With respect to purification, Akash, what are the bottlenecks that you see, and what are some of the potential solutions for dealing with those bottlenecks?
- Q (SS): In addition to purification, one of the primary challenges in gene therapy production is the overall high manufacturing cost. What are the primary reasons for this high cost and how can we start to bring them down?
- Q (SS): Where do you think the biggest value lies in terms of optimizing viral vector production? In other words, which steps in the workflow do you think have the most room for improvement?
- Q (SS): Production is just one part of the story, and characterization is the other. Akash, can you give us an overview of some of the most popular characterization techniques that are being employed in the gene therapy field?
- Q (SS): Leisha, can you comment on how characterization and analytics are involved in process development? Specifically, how far upstream can you take some of these techniques?
- Q (SS): Klaus, can you describe some of the major differences between in-line analytics during production compared to end-point analytics for the finished product?
- Q (SS): Akash – how do you qualify and validate an AUC method?
- Q (SS): What about higher-order species and aggregates?
- Q (SS): Leisha, can functional/transduction assays be done on AAVs obtained from crude culture, or does the sample need to be purified? If so, to what extent does the AAV need to be purified?
- Q (SS): Audrey, can you comment on the regulatory landscape, specifically in the context of the cGMP characterization?
- Q (SS): Klaus – can you comment on the state of the art for cGMP characterization that you use, and what you would like to see?
- Q (SS): Can each of you give me a summary on what you think the future of gene therapy looks like?
- Authorship & Conflict of Interest
- Article & copyright information