Understanding raw material performance: quality and consistency of cytokines for translation to the clinic
Cell & Gene Therapy Insights 2021; 7(10), 1147–1152
10.18609/cgti.2021.153
A deep understanding of origin, performance and quality of raw materials in cell and gene therapy development is crucial, but international standards for these raw materials are still missing in our industry. The challenge lies in the fact that raw biological materials are inherently variable, while batch-to-batch consistency is essential for successful and long-term commercialization of therapies. There is also the big debate of when to apply rigid standards, is commercial-scale manufacturing too late? One category of raw materials that require further standardization and characterization is cytokines, growth and differentiation factors (here named cytokines for simplicity). Cytokine quality and performance are directly linked to the clinical and commercial success of a therapy. However, there are important quality considerations to address during preclinical research to ensure your therapy is set up for regulatory and commercial success. Even cytokines that were originally developed for other uses, including those used as human therapeutics themselves, are not necessarily suited for use in cell and gene therapy manufacturing. Limited information on the potency and other critical attributes of the materials makes it difficult to define specifications for those reagents and to investigate the material. This article will explore ways of easing translation from preclinical development into the clinic, the importance of using animal-derived component-free (ADCF) cytokines, how to compare cytokines from different vendors and the value of international units of measurement.
Cytokine activity measurements & international standards
Defining and measuring the possible effects of a given cytokine is not a simple, black-and-white task; this is because of the inherent variable nature of these tiny but powerful messengers. Cytokines can impact a multitude of cells through the signaling pathways they initiate, in a wide variety of ways. Their effects can depend on various factors including the target cell, its environment, cell culture etc., adding another layer of variability to an already complex picture.
A cytokine’s biological activity should therefore be measured by its effect on a particular cell type. However, there is not yet a recognized industry standard for these measurements or their units, also because different protocols may be based on different modes of action of the same cytokine. This can be challenging for developers when trying to demonstrate reproducibility and comparability to regulatory authorities.
What we do have currently are ‘international units’ (IU) developed by the World Health Organization (WHO). IU are calculated using a standardized assay in which the cytokine of interest is tested side by side with the defined WHO standard, which can be obtained from the National Institute for Biological Standards and Control (NIBSC). The activity of the cytokine is then normalized using this standard. Such a standardized assay should in addition be validated following the applicable ICH guidelines, according to each lab’s specific conditions.
The NIBSC’s definition [1]NIBSC International Standards. of international units is:
“International units (IU) are assigned to international standards or other reference materials to allow the assessment of ‘biologicals’ in a consistent internationally agreed manner.
Biological reference materials, with an assigned value in IU, may be used in situations where physico-chemical determination of international standard units, e.g. mass, is not possible or not appropriate. There may be no agreed validated reference methods of determination available, or a simple mass unit may not adequately define a clinically-relevant measure of activity e.g. glycoprotein hormones.”
Examining the specifications for activity used by several leading cytokine providers indicates that IU are not broadly used by all suppliers. If these units were to be universally adopted as a global standard, it would allow the cell and gene therapy industry to:
Achieve comparability in the activity of cytokines in an internationally agreed manner and more easily evaluate cytokines from different raw material providers;
Produce comparable and reliable data, demonstrating batch-to-batch consistency for regulatory submission.
Batch-to-batch consistency is vital for regulatory approval and sustained commercial viability.
How to perform effective comparative side-by-side testing
There are many ways to measure the biological activity of cytokines, but quantifying all the potential activities of a cytokine in one single numeric value is not possible. We recommend measuring one defined effect – e.g. stimulation of cell proliferation – on one defined target cell under standardized conditions. The assays should be validated following ICH guidelines for each cytokine and should be performed according to SOPs under a GMP quality assurance system and using qualified equipment.
It’s important to be vigilant when comparing units of activity between different sources, as different suppliers may have their own methods for defining a unit. The method used must be the same between sources for a unit comparison to be valid. The only way to make a reliable comparison is via side-by-side testing. Unfortunately, this side-by-side testing can be arduous and time-consuming, here are some considerations to ensure effective testing.
Effective comparative side-by-side testing in three steps:
- Determine the activity in IU/mg: use identical proliferation assays and calibrate against an international reference standard;
- Determine the protein content in µg: use identical assays for all samples (the specific cytokine activity is dependent on the amount of cytokine tested). Note that different sources of international standard and test material may result in different molecular weights (e.g., caused by different amino acid sequences or different levels of glycosylation);
- Determine the purity: preferably using a HPLC method.
Why use cytokines free of animal- & human derived components?
Materials of biological origin, particularly of human or animal origin, can present risks, including transmission of adventitious agents or introduction of biological impurities.
Using cytokines free of any animal or human-derived components:
- Minimizes potential variables associated with animal or human-derived components
- Eliminates the risk of introducing biological contaminants in your cell and gene therapy process
- Speeds your time to market and saves money: no viral safety studies needed
To allow cell and gene therapy developers to perform a risk assessment of the raw materials used in their manufacturing process it is important that raw material suppliers offer a well-defined animal-derived component-free (ADCF) policy. ISO Technical Standard-20399 [2]ISO/TS 20399-1:2018 Biotechnology – Ancillary materials present during the production of cellular therapeutic products – Part 1: general requirements. ISO/TS 20399-2:2018 Biotechnology – Ancillary materials present during the production of cellular therapeutic products – Part 2: Best practice guidance for ancillary material suppliers. ISO/TS 20399-3:2018 Biotechnology – Ancillary materials present during the production of cellular therapeutic products – Part 3: Best practice guidance for ancillary material users. defines two ADCF levels:
- Level 1 (product level): the raw material does not contain any materials from animal or human source as its ingredients.
- Level 2 (production level): in addition to ADCF level 1, raw material is produced without the use of any materials from an animal or human source. This includes excipients, equipment or containers that come into contact with the raw material during production.
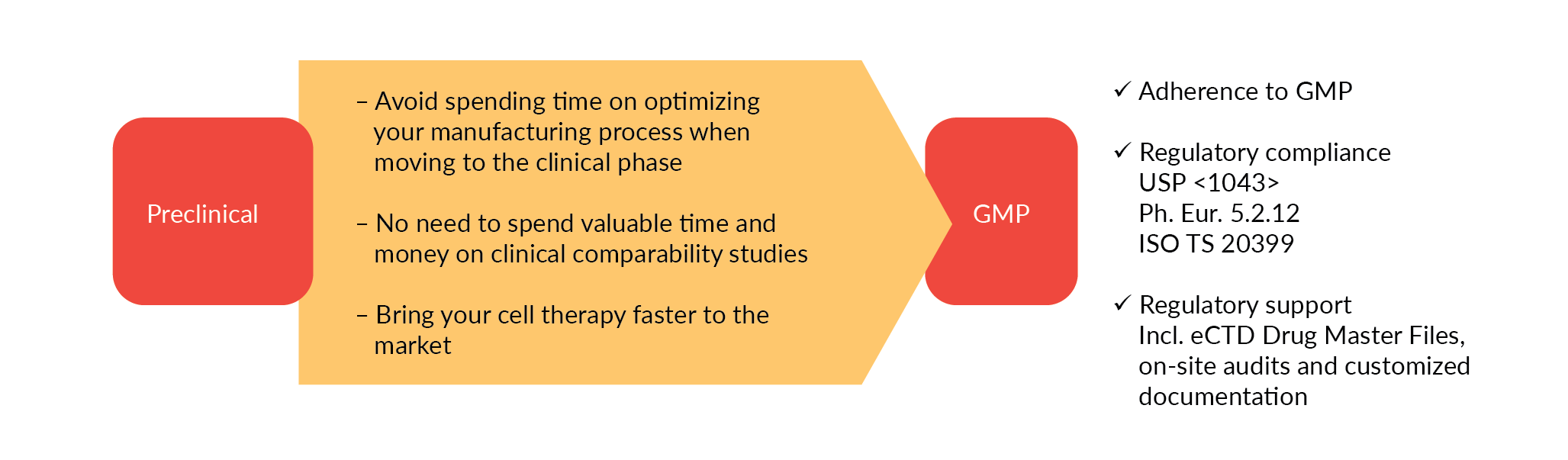
Seamless transition from preclinical to clinical development
Translation from lab to clinic and subsequent scale-up to commercial levels present our industry with many challenges, including reliability and reproducibility. GMP-grade raw materials that are required in later clinical phases may have different characteristics to those used in earlier research phases, as they are subject to different manufacturing protocols or quality standards. Changing raw materials during clinical development is time consuming and costly, requiring comparability studies that can have serious regulatory implications. Switching to GMP grade raw materials in the early clinical phase offers an economic benefit and saves time. A study from the Tufts Center for the Study of Drug Development (CSDD) estimated that the costs of an amendment in phase Ill are more than three times than those for a Phase 2 trial [3]Getz KA, Stergiopoulos S, Short M et al. The Impact of Protocol Amendments on Clinical Trial Performance and Cost. Ther. Innov. Regul. Sci. 2016; 50(4): 436–41..
To enable a safe and effective translation to the clinical stage we recommend using appropriately characterized cytokines of comparable performance in your preclinical and early development phase. These cytokines, we call them ‘preclinical grade’, should be produced under comparable conditions as the GMP equivalent, offering equal product performance. That way, you can switch directly to GMP-grade raw materials and avoid additional process optimization and laborious, expensive comparability studies (Figure 1
Conclusion
Characterizing and measuring the biological activity of cytokines is a critical factor in the clinical and commercial success of cell and gene therapies. International standards are crucial for comparability of cytokine activity and, therefore, batch-to-batch consistency and the industry must work together to develop and adopt standards.
The earlier and easier the switch to GMP grade materials can be made, the more cost and time effective it is. Ideally this will be done prior to the clinical phase to avoid comparability issues and potential regulatory hurdles.
References
1. NIBSC International Standards. Crossref
2. ISO/TS 20399-1:2018 Biotechnology – Ancillary materials present during the production of cellular therapeutic products – Part 1: general requirements. ISO/TS 20399-2:2018 Biotechnology – Ancillary materials present during the production of cellular therapeutic products – Part 2: Best practice guidance for ancillary material suppliers. ISO/TS 20399-3:2018 Biotechnology – Ancillary materials present during the production of cellular therapeutic products – Part 3: Best practice guidance for ancillary material users. Crossref
3. Getz KA, Stergiopoulos S, Short M et al. The Impact of Protocol Amendments on Clinical Trial Performance and Cost. Ther. Innov. Regul. Sci. 2016; 50(4): 436–41. Crossref
Affiliation
Bernd Leistler
Vice President Production, Sartorius CellGenix GmbH

Authorship & Conflict of Interest
Contributions: All named authors take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Acknowledgements: None.
Disclosure and potential conflicts of interest: The author declares that they have no conflicts of interest.
Funding declaration: The author received no financial support for the research, authorship and/or publication of this article.
Article & copyright information
Copyright: Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0 which allows anyone to copy, distribute, and transmit the article provided it is properly attributed in the manner specified below. No commercial use without permission.
Attribution: Copyright © 2021 Sartorius CellGenix. Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0.
Article source: Invited; externally peer reviewed.
Submitted for peer review: Sep 6 2021; Revised manuscript received: Sep 29 2021; Publication date: Oct 14 2021.