
Standardizing viral vector manufacture: maximizing production with the TRiP SystemTM
Cell Gene Therapy Insights 2018; 4(10), 983-994.
10.18609/cgti.2018.099
The use of viral vectors to create novel gene- and cell-based medicines is now a reality. As gene therapy matures into a new era, the industry will need to adopt improvements to viral vector manufacturing to meet the demand for GMP grade material. The activities of process optimisation/characterisation with any given viral vector entering into clinical development are considerable. The cell culture (Upstream) and purification/concentration (Downstream) aspects of this process are multi-faceted. The amount of vector produced during Upstream can vary depending on the transgene encoded, especially if the active protein is expressed in the production cell when constitutive or leaky tissue specific promoters are employed. Oxford BioMedica has developed the Transgene Repression In vector Production (TRiP) System™ to recover vector titers compromised by transgene expression. The system utilises the bacterial protein TRAP and its short RNA binding sequence – inserted within the transgene leader sequence – to repress transgene mRNA translation during vector production only, leaving expression unaffected in target cells. The TRiP System™has been used to fully recover titers of Lenti-, Adeno- and AAV-based vectors, and is expected to be universally applicable to any viral vector/vaccine platform. We anticipate that the TRiP System™ will enable new gene therapies to be considered, and assist those already in development to become commercially viable. Given that only TRAP and the viral proteins of the vector platform being employed will be expressed during Upstream, the TRiP System™ opens the door to ‘plug-and-play’ manufacturing, greatly minimising the burden of process development within a given pipeline.
The development of new gene therapies for genetic disorders and cancer is increasing at a rapid pace. Several promising therapies have recently received regulatory approval and are now commercialized, including Kymriah™ and Yescarta® for the treatment of certain B-cell malignancies, and Luxturna™ for the treatment of RPE65-mediated inherited retinal disease [1–3].
The contemporary vectors of choice are derived from engineered viruses including those based on gamma-retroviruses (RVs), lentiviruses (LVs), adeno-associated viruses (AAVs) and adenoviruses (AdVs), although a variety of non-viral vector approaches are now gaining greater momentum [4]. Given the diverse complexities associated with the broad range of indications being targeted by vectored gene therapies, it is likely that different viral vectors will ‘find their niche’ and that no single viral vector will become the de facto ‘industry standard’. As the successes of these approaches in clinical trials begin to build towards regulatory approval and commercialization, attention has focused on the emerging bottleneck in mass production of GMP grade vector material [5]. A way to overcome this challenge is to find new ways to maximize titer during viral vector production. Oxford BioMedica has developed the TRiP SystemTM, a new technology that allows suppression of the transgene expression in the production cell, essentially mitigating any side effects of the therapeutic protein on the manufacturing process. The TRiP SystemTM is on its way to become a new standard in viral vector manufacturing, implemented across diverse viral vector systems.
VIRUS TO VECTOR
The current break-through gene therapy products owe their success partly to the previous three decades of viral vector platform development, wherein complex virus genomes have been stripped down to their minimal functional sequences, and components separated onto multiple plasmids, to enable safe, efficient and stable delivery of transgenic cassettes to primary cells. In many cases, this has involved removal of various auxiliary and accessory genes specific to the virus/vector platform that are considered redundant in ‘single-round’ transduction-competent viral vectors [6–8]. Additionally, removal of certain functions, such as the multiple gene-regulator tat from HIV-1 based LVs, were important safety steps in which Oxford BioMedica played an important role [9]. Adenoviral vector development from first to third generations started with progressive deletion of regulatory and immune-regulatory genes (E1, E2, E3, E4) from the vector genome, and has essentially finished at completely ‘gutted’ AdVs, although these remain dependent on helper AdVs that are difficult to remove entirely from final vector material [10]. Similarly, AAV vectors have been engineered such that only the cis-acting viral sequences that allow production and packaging of the vector genome are present with transgenic sequences.  A key advancement in retroviral vector development was the ‘self-inactivating’ (SIN) modification to the vector genome to remove viral enhancer-promoter sequences, greatly limiting the chances activation of proto-oncogenes within the neighbouring chromosome [11].
A key advancement in retroviral vector development was the ‘self-inactivating’ (SIN) modification to the vector genome to remove viral enhancer-promoter sequences, greatly limiting the chances activation of proto-oncogenes within the neighbouring chromosome [11].
Together with sequence optimization processes, such as codon-optimization of packaging cassettes (minimizes recombination between components) [12], these modifications have led to the current state-of-art viral vector platforms in which no replicating competent entities have been detected to date [13–15]. Whilst there remains a theoretical risk of insertional mutagenesis by SIN-LVs, a realization of such an event has not been empirically observed in patients. In cases where only short term transgene expression is required, integration-defective LVs (ID-LVs) harboring mutation(s) in the integrase protein and/or in cis-acting elements on the vector genome are being used [16], as well as AAV vectors, which integrate into the host genome at low levels (~0.1%) [17]. Such rational design of viral vectors continues alongside the burgeoning field of ‘library-enhanced’ viral vectors, for example DNA shuffling of AAV capsid sequences for improved targeting and escape of pre-existing immunity [18]. Examples of rational design have led to the principle of targeting of enveloped viral vectors by pseudotyping with different viral glycoproteins [19], as well as microRNA-regulated vectors [20], and the development of novel vector architecture [21].
THE CHALLENGES OF VIRAL VECTOR PRODUCTION
Historically, the main choice of viral vector cell line has been those based on the HEK293 cell line developed by Frank Graham and colleagues several decades ago [22], and later the HEK293T cell line that expresses the SV40 large T antigen [23]. Effectively, researchers have selected these cells by trial-and-error to find that they typically yield higher titers than other cell lines. This is perhaps in part due to their high transfection efficiency but in hindsight, also due to their relatively low (or absent) expression levels of viral restriction factors that have subsequently been identified by the research community over the last 15 years [24]. Only relatively recently have other cell lines been developed to provide alternative base cell lines for vector production [25,26]. Whatever the choice of production cell, the output of viral vector titers during the ‘Upstream’ process phase can be affected by several different factors, for example [27]:
- Viral serotype/pseudotype employed;
- Transgenic sequence composition and size;
- Media composition/gassing/pH;
- Transfection reagent/process;
- Chemical induction and vector harvest timings;
- Cell fragility/viability;
- Bioreactor shear-forces; and,
- Impurities
The potential variability in the composition of ‘crude’ harvest material typically impacts on the performance of the different steps taken during the ‘Downstream’ process, for example ion-exchange chromatography, size-exclusion and (sterile) filtration steps [28]. Both Upstream and Downstream processes ultimately determine the purity of the drug product (DP) being administered. Certain gene therapies for direct in vivo administration will likely require high DP titers in order to achieve target doses. This requires large concentration factors over the process, and if crude harvest titer is limiting this will necessarily result in ‘over’-concentration of impurities such as residual DNA.

However, one single factor that affects many of these considerations is the potential expression of the transgene product during the Upstream process. This often means that the entire viral vector process will be bespoke for a given transgene-expressing vector, which places a considerable burden of resource and time for process characterization and validation.
HIGH TRANSGENE EXPRESSION: BE CAREFUL WHAT YOU WISH FOR
The level of transgene expression within vector production cells is dictated by several cis-acting nucleotide sequences designed into the expression cassette: promoter/enhancer elements (transcription activity) [29]; introns/polynucleotide length-composition/polyadenylation (mRNA export/stability) [30,31]; and Kozak sequence/UTR length-composition/codon usage (translation efficiency) [32–34]. Clearly, there are other factors in the make-up of the protein itself that contribute to its half-life and activity; indeed, it is protein activity that is ultimately the most important factor both from the perspective of in vivo efficacy and the unwanted activity during vector production. Typically, the researcher will aim to optimize all of these sequences in order to maximize activity in the target cell to increase chance of therapeutic benefit and to potentially allow lower dosing levels. However, unwanted expression during vector production often remains as a ‘brut fact’ after this optimization process. The impact of transgene activity during viral vector production can sometimes be predicted from the outset (such as a known cytotoxic protein) but others cannot (Figure 1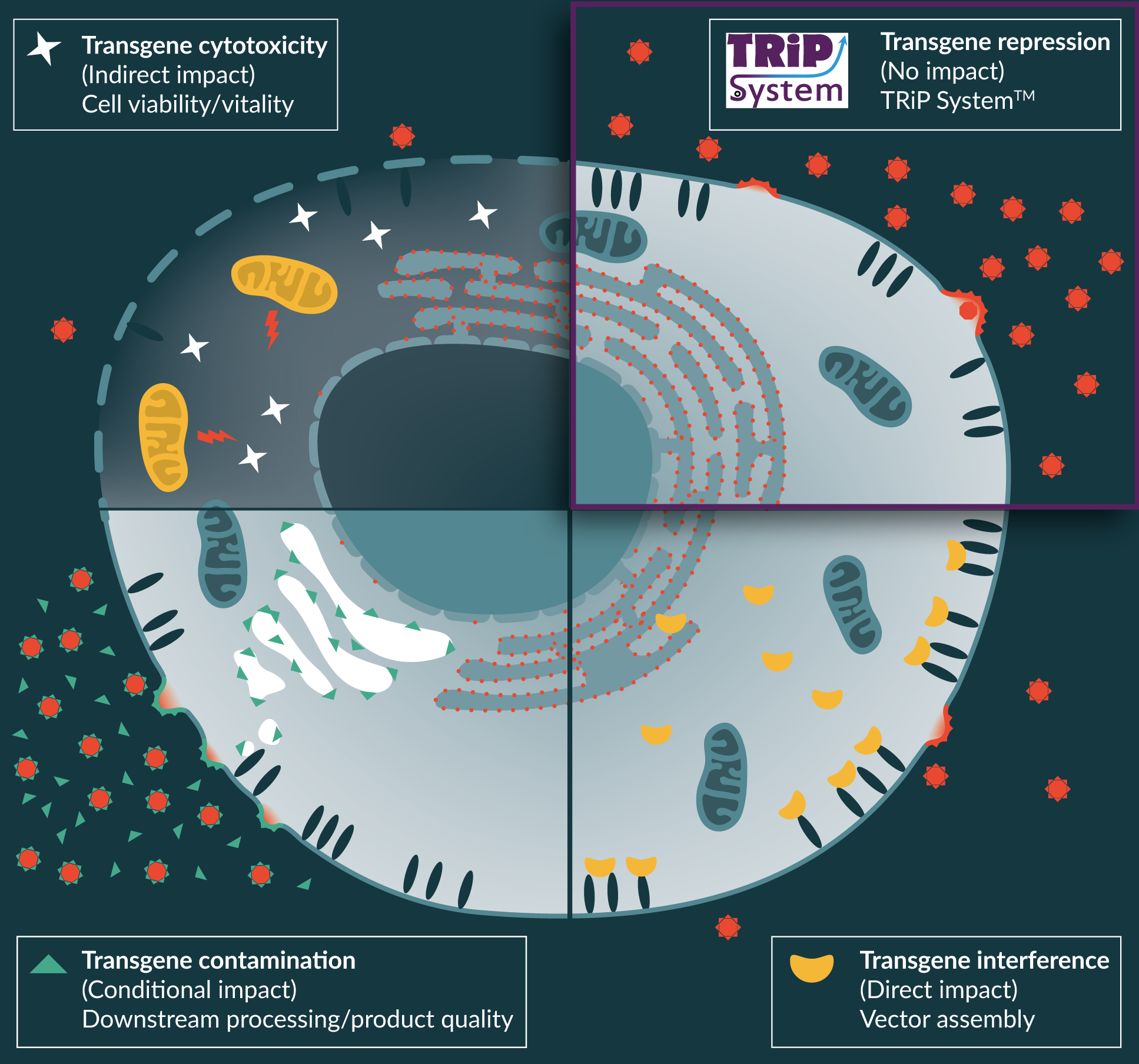
It is usual, when conducting early proof-of-efficacy experiments in pre-clinical models to consider 3-to-5 fold lower vector titers (compared to benchmark values) as practicable levels of vector production at research scales, without contemplating the knock-on consequences for scale-up and cost-of-goods at GMP production scales. This ultimately may determine whether a promising therapy can be realized commercially. Still other conceptually promising approaches may be terminated during these early development stages because the transgene activity is so potently detrimental to viral vector titers. Therefore, the ability to repress transgene protein expression/activity during vector production becomes extremely desirable. To do so may allow previous viral vector therapies – once hampered by poor titers – to be fully evaluated for clinical development. It would also represent a significant step to standardized viral vector manufacturing processes whereby only the viral vector components are expressed during the Upstream stage.
TRANSGENE: QUIET PLEASE!
There are a number of approaches to minimize transgene expression during viral vector production. The development of next generation tissue-specific promoters is increasing within the gene therapy field. Whilst minimizing transgene expression within vector production cells is achievable with use of certain tissue specific promoters, leaky expression is possible and will always need to be determined empirically. For example, the Adenovirus E1a and SV40 Large T gene products expressed in HEK293T cells have been shown to be promiscuous transactivators of housekeeping and tissue-specific genes [37–40]. Current manufacturing methods that require large quantities of plasmid DNA per cell at transfection may result in a substantial amount of transgene expression from even very ‘quiet’ tissue specific promoters. The initial cost and time investment in order to develop a bespoke tissue specific promoter (and for it to be validated in all the specific models of choice in pre-clinical studies) may be too great to justify, especially when there may be proven alternatives immediately available. Moreover, the validation of specificity/activity of a bespoke promoter within animal models may not necessarily translate in the clinic. Additionally, there are settings in which the use of completely silent tissue specific promoters will not quench transgene expression. For example, we have shown that the genomic RNA of RVs/LVs is a proficient mRNA for translation of ORFs directed by IRES elements, leading to substantial transgene expression even when the transgene cassette has no promoter [41]. Finally, it actually may be desirable to use strong constitutive promoters to maximise expression in a wide number of target tissues if such an approach is required to realize clinical efficacy.
Other molecular tools available that can achieve various degrees of transgene repression include: tet/cumate ON/OFF and tetR/lacR repressor systems (transcription control) [42–45], ribozyme switches/microRNA (mRNA stability) [46] and protein-degron systems (protein stability) [47,48]. The caveats to such approaches typically fall into at least one of four categories: difficult to engineer (e.g., inserting tet-operator sequences into promoter of choice without compromising promoter functionality); modest levels of repression achieved; impact on vector utility (titers, capacity); and risk of non-human protein sequences being expressed or appended to the transgene protein in vivo.
TRANSGENE REPRESSION IN VECTOR PRODUCTION (TRiP) SYSTEM™
Oxford BioMedica recently reported the development of the TRiP SystemTM, a universal transgene repression system for limiting translation of transgene ORFs during viral vector production [49]. The TRiP SystemTM is based on previous characterisation of the bacterial protein ‘tryptophan RNA-binding attenuation protein’ (TRAP) [50]. In bacteria such as bacillus subtilis, TRAP is a component of the L-tryptophan (L-trp) synthase operon feed-back loop [51].  TRAP is a small protein of ~8kDa in size, which self-assembles into a toroidal structure composing typically 11 monomers. The resulting tyre-like scaffold contains 11 L-trp-binding pockets, which when bound by L-trp causes a conformational change such that TRAP binds to its target RNA sequence around the ‘tyre tread’. The trp operon is regulated by TRAP both at the level of pre-mature RNA polymerase termination and at the level of translation initiation via binding to the TRAP binding sequence (tbs) within the RNA leader of a number of genes in the operon.
TRAP is a small protein of ~8kDa in size, which self-assembles into a toroidal structure composing typically 11 monomers. The resulting tyre-like scaffold contains 11 L-trp-binding pockets, which when bound by L-trp causes a conformational change such that TRAP binds to its target RNA sequence around the ‘tyre tread’. The trp operon is regulated by TRAP both at the level of pre-mature RNA polymerase termination and at the level of translation initiation via binding to the TRAP binding sequence (tbs) within the RNA leader of a number of genes in the operon.
In the TRiP SystemTM, the tbs is inserted close to the initiation codon of the transgene of interest, and the resulting stable TRAP-tbs complex that forms upstream results in inhibition of translation of the protein (Figure 2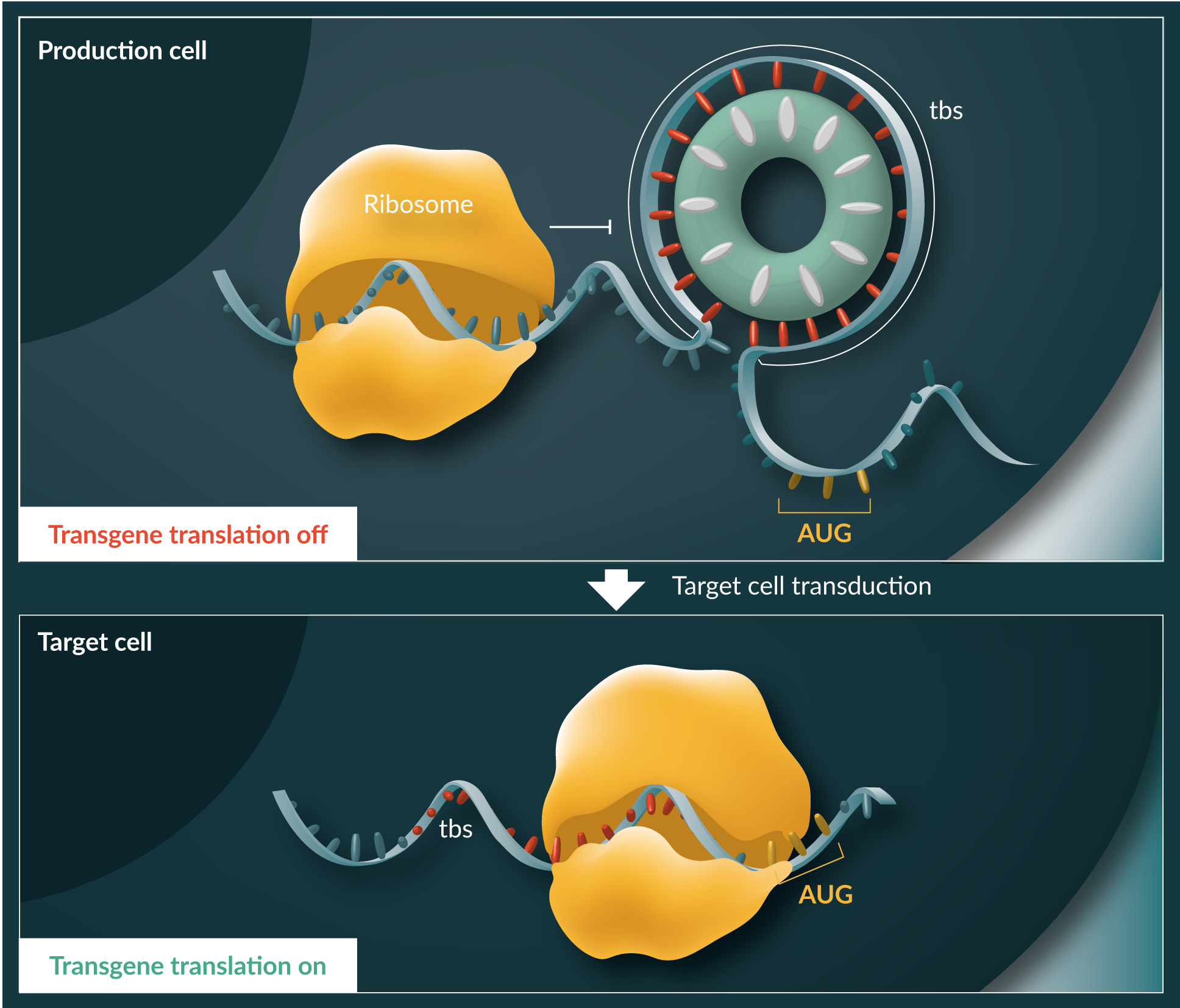
Importantly, the simple components of the TRiP SystemTM do not appear to impact on the fundamental biology of the classic vector platforms, namely LVs, AAV and AdV vectors. In principle it could be applied to other viral vectors and ‘armed’ vaccines. Of further importance is the ability to produce stable cell lines constitutively expressing the TRAP protein, indicating that there is no obvious cytotoxicity associated with this RNA-binding protein. At Oxford BioMedica we have recently leveraged a bespoke, automated high through-put cell line isolation/screening platform to isolate serum-free, suspension TRAP-expressing cell lines that maintain high levels of transgene repression over several weeks.
The TRiP SystemTM enhances output titers of LVs encoding CARs, as well as fully rescuing AAV and AdVs expressing pro-apoptotic factors to benchmark levels [49]. Table 1 presents examples of the levels of recoveries in vector titers capable in leveraging the TRiP SystemTM across different platforms. Further benefits of the TRiP SystemTM were exampled by evaluation of an LV vector constitutively expressing the Cyclo-oxygenase-2 (COX-2) gene under control of the CMV promoter (for treatment of Glaucoma). This vector typically produces up to 1000 times lower vector titers compared to the GFP control. The TRiP SystemTM rescued vector output of the LV-CMV-COX-2 vector to the same levels as the GFP vector, and remarkably also improved the protein profile of the concentrated LV material [49]. The high expression of COX-2 during LV production had a broad impact on the abundance and type of host cell proteins associated with concentrated LV particles, including the amount of VSVG envelope protein incorporated. Repressing COX-2 expression resulted in a similar protein profile to that of a GFP-expressing LV, as well as rescuing VSVG-incorporation into virions.
| Table 1. Examples of typical viral vector titer recoveries when utilizing the TRiP SystemTM. | ||
|---|---|---|
| Platform | Transgene/vector | Titer recovery |
| TRiPLenti | Cox-2/EIAV | 600-fold |
| TRiPLenti | FVIII/EIAV | 10-fold |
| TRiPLenti | FPR/EIAV | 10-fold |
| TRiPLenti | VEGF-B/EIAV | 10-fold |
| TRiPLenti | CAR/HIV-1 | 30-fold |
| TRiPAAV | Bax/scAAV2 | >10-fold* |
| TRiPAdeno | Bax-IRES-GFP/Ad5 | 100,000-fold |
| The TRiP SystemTM is applicable to many viral vector platforms, including Lenti-, Adeno-and AAV-based vectors. The table presents observed increases in vector titers across these three vector platforms using different transgenes, all expressed by the potent CMV promoter [49]. Transgenes: Cyclooxygenase-2, Factor VIII, Prostaglandin receptor (FPR), Vascular endothelial growth factor B, a Chimeric antigen receptor, Bcl-2-associated X. *Likely underestimate due to assay LOQ. | ||
The protein profiling analysis also revealed that the TRAP protein itself is of high abundance within LV material, presumably due to passive incorporation of this abundant cytoplasmic protein into virions during the budding process. Clearly this initial finding represented an open question as to whether the presence of TRAP protein with LV material might compromise a direct in vivo administration gene therapy approach. Given that re-dosing of patients with LVs is not likely to be required (and may not be possible given the likely immune responses raised to vector envelopes such as VSVG [53]), the most likely theoretical hindrance to gene delivery by LVs produced using the TRiP SystemTM would be pre-existing antibody responses to TRAP protein. Bacillus is a genus of Gram-positive, rod-shaped bacteria found in soil and water [54], and so it’s highly likely that most humans will have been exposed to this organism from a young age. However, it is also a gut-commensal organism suggesting that humans may have been generally tolerised [55]; such pre-existing immune responses to TRAP would therefore be unlikely. In our initial work, no immune response to TRAP was detected when evaluating the LV-COX-2 vector in a small rat study [49].

To empirically determine if such responses exist in humans, we have subsequently initiated screening of patient sera for pre-existing antibody responses to the TRAP protein. Within the limits of sensitivity of the assay employed, we have not detected antibody responses from 25 patient sera to native or non-native TRAP, either in its 11-mer or monomeric form [Unpublished Data]. It is of note that similar techniques employed to determine pre-existing antibody responses to Cas9 protein, detected responses in 79% of donors probing against SaCas9 (S. aureus) and 65% of donors probing against SpCas9 (S. pyogenes) [56]. Whilst later reports using assays based on ELISA have suggested that more accurate response rates to Cas9 may be lower at ≤10% [57], the lack of any demonstrable antibody responses to TRAP in our ongoing study already suggests that the presence of TRAP protein within viral vector material will not be an issue for the general population.
TAKING A TRiP TO THE LAB
It is exciting to see other researchers now start to evaluate the The TRiP SystemTM for use in generation of ‘difficult’ vectors. For example, the Jenner Institute in Oxford is developing novel vaccination approaches against a wide range of pathogens, such as Malaria, Influenza, Ebola and HIV-1, using the simian (chimpanzee) adenovirus (ChAdV) vector platform in combination with modified vaccinia virus Ankara (MVA). Professor Tomáš Hanke, one of the collaborators in the TRiPAdeno evaluation, is taking an approach to develop HIV-1 vaccines by designing novel immunogens composed of multiple, M-group conserved regions of the virus, and has already reported some promising results [58]. However, production of the recombinant ChAdV vectors expressing these highly artificial proteins has proven to be difficult in some cases, and the TRiP SystemTM is giving new hope:
Professor Tomáš Hanke, Jenner Institute, University of Oxford, UK: The HIV-1 based immunogens that we are developing are bioinformatics-informed, computed chimeric proteins derived from the most conserved regions of the viral proteome and are of over 700 amino acids in length. These can be expressed with or without the tPA-leader secretory sequence. We have found that ChAdVs expressing these types of protein mosaics, which do not have any natural folding, can destabilize the recombinant vector during amplification. In other contexts where transgene ORFs are stable, we find that downstream processing steps can also be differentially affected depending on the transgene encoded. As a result, the recombinant vectors may be hard to rescue and sequencing of the transgene ORF within the vector genomes may reveal small nucleotide insertions/deletions causing a shift in the reading frame, expression of irrelevant amino acids and premature termination. This has previously led us to employ the tetR repression system, which in some cases has proven successful. However, we have found that even using the tetR system, some recombinant vectors expressing certain difficult products can be hard to rescue due to the leakiness of the tetR repression. This goes to show how unpredictable the impact of expression of some of these proteins can be on Adenoviral vector biology. We’re therefore enthusiastic about the ongoing collaboration with Oxford BioMedica to evaluate the TRiP SystemTM in production of recombinant ChAdVs, and our initial results look promising.
Still others have leveraged the TRiP SystemTM as a helpful research tool:
Professor Greg Towers, University College London: Our goal was to understand whether a particular transgene was responsible for low lentiviral vector production efficiencies; was the transgene causing cytotoxicity, and reducing vector output from the producer cells? The TRiP SystemTM allowed us to control transgene expression in the transfected HEK293T cells. We used a tbs-containing transgene cassette, and tested a GFP LV genome in parallel as a control. Co-expression of TRAP effectively switched off GFP and our transgene expression, allowing us to clearly show that in this case transgene expression itself was not responsible for poor vector production. We eventually worked out that another feature of the transgene RNA was responsible, allowing us to fix this through further construct design. Overall, the TRiP SystemTM was a tractable and straightforward system for repression transgene expression in producer cells, and we were very pleased with how it worked.
We anticipate that the TRiP SystemTM will be employable in other areas of research, such as expanding the complexity in vectored cDNA libraries, especially those encoding novel modified or synthetic proteins. Typically, vectored libraries are produced by transient transfection of vector production cells with a complex mix of DNA comprising the entire cloned/synthesized library; each vector genome expressing an individual clone. Given that each production cell will likely be transfected with multiple plasmids, then the expression of just one of these clones expressing a gene that impacts on cell viability/vitality or vector assembly will compromise the production of all the other clones transfected into that cell. Currently, the only way to mitigate this problem is to massively up-scale production of the vector library to ensure that ‘somewhere’ within the transfected culture every vectored clone is produced from cells that are not expressing a problematic gene. Therefore repression of apoptotic, toxic or dominant-negative genes using the TRiP SystemTM during the initial production of the viral vector library will expedite the production of maximal library complexity that would otherwise be limited by the activities of those proteins.
In summary, the TRiP SystemTM is fundamentally an approach to rescue or restore viral vectors at or close to the ‘benchmark’ titers achievable with any given viral vector platform, and could be of immediate benefit to researchers programs already in clinical development. However, its application will also help to prevent early stage gene therapy product failures related to transgene protein expression, and also represents the first steps towards ‘plug-and-play’ viral vector manufacture within commercial pipelines, avoiding continual process development for each new product. The TRiP SystemTM will pave the way to faster and more consistent translation of gene therapy strategies from theory to clinical success.
Financial & Competing Interests Disclosure
Dan Farley is an employee of Oxford BioMedica, which has funded the subject matter and materials discussed in this manuscript. He also has stock ownership and options in the Company and also holds patents and patents pending. No writing assistance was utilized in the production of this manuscript.
References
1. Leahy AB, Elgarten CW, Grupp SA, Maude SL, Teachey DT. Tisagenlecleucel for the treatment of B-cell acute lymphoblastic leukemia. Expert Rev. Anticancer Ther. 2018; 18(10): 959–71. CrossRef
2. Ghobadi A. Chimeric antigen receptor T cell therapy for non-Hodgkin lymphoma. Curr. Res. Transl. Med. 2018; 66(2): 43–9. CrossRef
3. Ameri H. Prospect of retinal gene therapy following commercialization of voretigene neparvovec-rzyl for retinal dystrophy mediated by RPE65 mutation. J. Curr. Ophthalmol. 2018; 30(1): 1–2. CrossRef
4. Chira S, Jackson CS, Oprea I et al. Progresses towards safe and efficient gene therapy vectors. Oncotarget 2015; 6(31): 30675–703. CrossRef
5. Van der Loo JCM, Wright JF. Progress and Challenges in Viral Vector Manufacturing.” Hum. Mol. Genetics 25 2016; R1: R42–R52. CrossRef
6. Dormond E, Perrier M, Kamen A et al. From the first to the third generation adenoviral vector: what parameters are governing the production yield? Biotechnol. Adv. 2009; 27(2): 133–44. CrossRef
7. Durand S, Cimarelli A. The Inside Out of Lentiviral Vectors. Viruses 2011; 3(2): 132–59. CrossRef
8. Naso MF, Tomkowicz B, Perry WL 3rd, Strohl WR. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. Biodrugs 2017; 31(4): 317–34. CrossRef
9. Kim VN, Mitrophanous K, Kingsman SM, Kingsman AJ. Minimal Requirement for a Lentivirus Vector Based on Human Immunodeficiency Virus Type 1. J. Virol. 1998; 72(1): 811–6. CrossRef
10. Dormond E, Perrier M, Kamen A. From the first to the third generation adenoviral vector: what parameters are governing the production yield? Biotechnol. Adv. 2009; 27(2): 133–44. CrossRef
11. Yu SF, von Rüden T, Kantoff PW et al. Self-inactivating retroviral vectors designed for transfer of whole genes into mammalian cells. Proc. Natl Acad. Sci. USA 1986; 83(10): 3194–8. CrossRef
12. Kotsopoulou, E, Kim VN, Kingsman AJ, Kingsman SM, Mitrophanous KA. A Rev-Independent Human Immunodeficiency Virus Type 1 (HIV-1)-Based Vector That Exploits a Codon-Optimized HIV-1 gag-Pol Gene. J. Virol. 2000; 74(10): 4839–52. CrossRef
13. Farley DC, Bannister R, Leroux-Carlucci MA, Evans NE, Miskin JE, Mitrophanous KA. Development of an equine-tropic replication-competent lentivirus assay for equine infectious anemia virus-based lentiviral vectors. Hum. Gene Ther. Methods 2012; 23(5): 309–23. CrossRef
14. Tareen SU, Nicolai CJ, Campbell DJ et al. A Rev-Independent gag/pol Eliminates Detectable Psi-Gag Recombination in Lentiviral Vectors. BioResearch Open Access 2013; 2.6: 421–30. CrossRef
15. Farley DC, McCloskey L, Thorne BA et al. Development of a replication-competent lentivirus assay for dendritic cell-targeting lentiviral vectors. Mol. Ther. Methods Clin. Dev. 2015; 2: 15017. CrossRef
16. Albershardt TC, Campbell DJ, Parsons AJ, Slough MM, Ter Meulen J, Berglund P. LV305, a dendritic cell-targeting integration-deficient ZVex(TM)-based lentiviral vector encoding NY-ESO-1, induces potent anti-tumor immune response. Mol. Ther. Oncolytics 2016; 3: 16010. CrossRef
17. Deyle, DR, Russell DW. Adeno-Associated Virus Vector Integration. Curr. Opin. Mol. Ther. 2009; 11.4: 442–7. CrossRef
18. Michelfelder S, Trepel M. Adeno-associated viral vectors and their redirection to cell-type specific receptors. Adv. Genet. 2009; 67: 29–60. CrossRef
19. Joglekar AV, Sandoval S. Pseudotyped Lentiviral Vectors: One Vector, Many Guises. Hum. Gene Ther. Methods 2017; 28(6): 291–301. CrossRef
20. Geisler A, Fechner H. MicroRNA-regulated viral vectors for gene therapy. World J. Exp. Med. 2016; 6(2): 37–54. CrossRef
21. Vink CA, Counsell JR, Perocheau DP et al. Eliminating HIV-1 Packaging Sequences from Lentiviral Vector Proviruses Enhances Safety and Expedites Gene Transfer for Gene Therapy. Mol. Ther. 2017; 25(8): 1790–1804. CrossRef
22. Graham FL, Smiley J, Russell WC, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977; 36(1): 59–74. (1977). CrossRef
23. DuBridge RB, Tang P, Hsia HC, Leong PM, Miller JH, Calos MP. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell Biol. 1987; 7(1): 379–87. CrossRef
24. Chemudupati M, Kenney AD, Bonifati S et al. From APOBEC to ZAP: Diverse mechanisms used by cellular restriction factors to inhibit virus infections. Biochim. Biophys. Acta Mol. Cell Res. 2018; pii: S0167-4889(18)30416-6. CrossRef
25. Fallaux FJ, Bout A, van der Velde I et al. New helper cells and matched early region 1-deleted adenovirus vectors prevent generation of replication-competent adenoviruses. Hum. Gene Ther. 1998; 9: 1909–17. CrossRef
26. Wölfel J, Ruth Essers, Corinna Bialek, Sabine Hertel, Nadine Scholz-Neumann, Gudrun Schiedner. CAP-T cell expression system: a novel rapid and versatile human cell expression system for fast and high yield transient protein expression. BMC Proc. 2011; 5(Suppl. 8): P133. CrossRef
27. Merten O-W, Schweizer M, Chahal P, Kamen AA. A. Manufacturing of viral vectors for gene therapy: part I. Upstream processing. Pharmaceut. Bioprocess 2014; 2: 183–203. CrossRef
28. Merten O-W, Merten O-W, Schweizer M, Chahal P, Kamen AA. A. Manufacturing of viral vectors for gene therapy: part II. Downstream processing and safety aspects. Pharmaceut. Bioprocess 2014; 2: 237–51. CrossRef
29. Papadakis ED, Nicklin SA, Baker AH, White SJ. Promoters and control elements: designing expression cassettes for gene therapy. Curr. Gene Ther. 2004; 4(1): 89–113. CrossRef
30. Valencia P, Dias AP, Reed R. Splicing promotes rapid and efficient mRNA export in mammalian cells. Proc. Natl Acad. Sci. USA 2008; 105: 3386–91. . CrossRef
31. Proudfoot NJ. Ending the message: poly(A) signals then and now. Genes Dev. 2011; 25(17): 1770–82. CrossRef
32. Polychronakos C. Gene expression as a quantitative trait: what about translation? J. Med. Genet. 2012; 49(9): 554–7. CrossRef
33. Leppek K, Das R, Barna M. Functional 5’ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2018; 19(3): 158–74. CrossRef
34. Hanson G, Coller J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 2018; 19(1): 20–30. CrossRef
35. Sanjana NE, Shalem O, Zhang F. Improved Vectors and Genome-Wide Libraries for CRISPR Screening. Nat. Methods 2014; 11(8): 783–4. CrossRef
36. Radcliffe PA, Sion CJ, Wilkes FJ et al. Analysis of factor VIII mediated suppression of lentiviral vector titres. Gene Ther. 2008; 15(4): 289–97. CrossRef
37. Zhao H, Granberg F, Elfineh L, Pettersson U, Svensson C. Strategic Attack on Host Cell Gene Expression during Adenovirus Infection. J. Virol. 77(20): 11006–15. CrossRef
38. Fukamizu A, Sagara M, Sugiyama F, Yagami K, Murakami K. Tissue-specific trans-activation of renin gene by targeted expression of adenovirus E1A in transgenic mice. Biochem. Biophys. Res. Commun. 1994; 199(1): 183–90. CrossRef
39. Gruda MC, Zabolotny JM, Xiao JH, Davidson I, Alwine JC. Transcriptional activation by simian virus 40 large T antigen: interactions with multiple components of the transcription complex. Mol. Cell. Biol. 1993; 13(2): 961–9. CrossRef
40. Cantalupo PG, et al. Cell-type Specific Regulation of Gene Expression by Simian Virus 40 T antigens. Virology 386(1):183-191. (2009) CrossRef
41. Supplementary information of ref 49 CrossRef
42. Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl Acad. Sci. USA 1992; 89(12): 5547–51. CrossRef
43. Cottingham MG, Carroll F, Morris SJ et al. Preventing Spontaneous Genetic Rearrangements in the Transgene Cassettes of Adenovirus Vectors. Biotechnol. Bioeng. 2012; 109(3): 719–28. CrossRef
44. Mullick A, Xu Y, Warren R et al. The cumate gene-switch: a system for regulated expression in mammalian cells. BMC Biotechnol. 2006; 6: 43. CrossRef
45. Hu MC, Davidson N. The inducible lac operator-repressor system is functional in mammalian cells. Cell 1987; 48(4): 555–66. CrossRef
46. Strobel B, Klauser B, Hartig JS, Lamla T, Gantner F, Kreuz S. Riboswitch-mediated Attenuation of Transgene Cytotoxicity Increases Adeno-associated Virus Vector Yields in HEK-293 Cells. Mol. Ther. 2015; 23(10): 1582–91. CrossRef
47. Neklesa TK, Winkler JD, Crews CM. Targeted protein degradation by PROTACs. Pharmacol. Ther. 2017; 174: 138–44. CrossRef
48. Chung HK, Jacobs CL, Huo Y et al. Tunable and reversible drug control of protein production via a self-excising degron. Nat. Chem. Biol. 2015; 11(9): 713–20. CrossRef
49. Maunder HE, Wright J, Kolli BR et al. Enhancing titres of therapeutic viral vectors using the transgene repression in vector production (TRiP) system. Nat. Commun. 2017; 8: 14834. CrossRef
50. Nie M, Htun H. Different modes and potencies of translational repression by sequence-specific RNA-protein interaction at the 5¢-UTR. Nucleic Acids Res. 2006; 34: 5528–40. CrossRef
51. Gollnick P, Babitzke P, Antson A, Yanofsky C. Complexity in regulation of tryptophan biosynthesis in Bacillus subtilis. Annu. Rev. Genet. 2005; 39: 47–68. CrossRef
52. Elliott MB, Gottlieb PA, Gollnick P. Probing the TRAP-RNA interaction with nucleoside analogs. RNA 1999; 5(10): 1277–89. CrossRef
53. Palfi S, Gurruchaga JM, Ralph GS et al. Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson’s disease: a dose escalation, open-label, phase 1/2 trial. Lancet 2014; 383: 1138–46. CrossRef
54. Traxler MF, Kolter R. Natural products in soil microbe interactions and evolution. Nat. Prod. Rep. 2015; 32(7): 956–70. CrossRef
55. Chistiakov DA, Bobryshev YV, Kozarov E, Sobenin IA, Orekhov AN. Intestinal Mucosal Tolerance and Impact of Gut Microbiota to Mucosal Tolerance. Front. Microbiol. 2014; 5: 781. CrossRef
56. Charlesworth CT, Deshpande PS, Dever DP et al. Identification of pre-existing adaptive immunity to Cas9 proteins in humans. bioRxiv 2018; doi:10.1101/243345. CrossRef
57. Simhadri VL, McGill J, McMahon S et al. Prevalence of Pre-existing Antibodies to CRISPR-Associated Nuclease Cas9 in the USA Population. Mol. Ther. Methods Clin. Dev. 2018; 10: 105–12. CrossRef
58. Ondondo B, Murakoshi H, Clutton G et al. Novel Conserved-region T-cell Mosaic Vaccine With High Global HIV-1 Coverage Is Recognized by Protective Responses in Untreated Infection. Mol. Ther. 2016; 24(4): 832–42. CrossRef
Affiliations
Dan Farley, PhD
Vector Engineering Senior Manager at Oxford BioMedica
![]()