A Novel Low Cost High Yield Clinical Scale Cell Separator
Cell Gene Therapy Insights 2018; 4(7), 581-600.
10.18609/cgti.2018.057
Submitted for Peer Review: Jun 25 2018 Published: Aug 22 2018
Introduction
Immunomagnetic cell separation (IMCS) is on the verge of playing a critical role in clinical therapies for cancers and genetic diseases. Seminal work by Molday introducing magnetic affinity separations to biological systems [1], his subsequent synthesis of targetable, colloidal, magnetic dextran nanoparticles [2], and Owen’s work introducing high-gradient magnetic separation (HGMS) to cell separation [3] laid the groundwork for IMCS. Kemshead and Ugelstad’s work on monodisperse magnetic polymer particles and their applications to cells ushered in the therapeutic utility of IMCS [4]. Miltenyi and colleagues perfected both Molday’s nanoparticles and HGMS [5], making their combination immensely useful for applications from the laboratory to the clinic. The evolution of commercial magnetic cell separation instruments and reagents has been reviewed by Zborowski [6], another pioneer in the IMCS field. These discoveries and innovations have played major roles in our understanding of cell biology and continue to play a key role in the manufacture of cell and gene therapeutic products for cancer and hereditary diseases.
Among the most exciting and most significant therapies coming online are those that utilize a patient’s immune system and advances in genetic engineering to create chimeric antigen receptor (CAR) T cells that attack tumor cells. Some remarkable and ostensibly sustained cures have recently been achieved in several hematology-related cancers. Those therapies and the likelihood of their extension to solid tumor therapies that will follow are based on a set of ingenious tools that make them possible. Adoptive cell transfer therapy with CAR-engineered T cells involves:
- Isolation of T cells from peripheral blood mononuclear cells (PBMC)
- Modification and reprogramming of T cells; and
- Transferal of T cells back into the patient.
These steps must be performed with the utmost precision and reliability; however, efforts should be made to reduce costs such that these therapies are not prohibitively expensive.
The tool of choice for purifying cells from leukapheresis products containing approximately 1010–1011 PBMC is immunomagnetic separation. At present, the clinical-scale cell separation market is dominated by Miltenyi Biotec GmbH with their weakly magnetic nanoparticles requiring steel-bead-packed columns to induce ultra-high magnetic gradients for collection; to a lesser extent Dynal technology employing 1–2.5 µm magnetic beads is used as well. One concern for users of Miltenyi’s system is the expensive disposable magnetic columns required and the possibility such columns can clog. Nevertheless Miltenyi has continued to add peripherals to their column-based system with very considerable success. On the other hand Dynabeads are highly magnetic, making them easy to separate; however, they can have harmful effects on cells. Both technologies are nearly 40 years old and given the potential promise that cell therapy affords, significantly improved methods will be required as this field evolves.
Highly magnetic colloidal nanoparticles like those introduced by Liberti et al. [7-16], which are referred to as ferrofluids (FF), have been commercialized in the CELLSEARCH® system–the only FDA-approved system for detecting circulating tumor cells (CTC). The CELLSEARCH® system has played a seminal role in the development of the CTC field, and FF, which are critical to the function of the CELLSEARCH® system, are particularly suitable for rare-cell isolation. For many of the same reasons FF are the ideal immunomagnetic nanoparticle for large-scale cell separations because they:
- Are the most magnetic biologically active nanoparticles by mass and, as such, do not require complex and costly ultra-high magnetic gradient columns to separate cells;
- Move with Brownian motion for quick reactions without the need for mixing;
- Are non-toxic to cells in culture as well as in vivo [17], [18];
- Are inexpensive to manufacture and can be filter sterilized;
- Are robust and have been market tested for CTC isolation in the CELLSEARCH® System; and
- Allow for process manipulations that no other system affords.
Hence FF are similar to Molday nanoparticles (used in Miltenyi Biotec’s CliniMACS instruments) in many respects including their binding efficiency and non-toxicity, but FF-labeled cells are considerably easier to separate because of their significantly larger magnetic moment. This key difference permits cell separations in a variety of vessels with relatively simple external-gradient magnetic separators, which has major implications for clinical applications. In particular, expensive single-use high-gradient magnetic columns–with their cell recovery and cell damage issues–need not be employed; instead, cells can be separated within inexpensive, disposable, sterile vessels–translating to a huge cost savings for the user. Moreover, the version of FF used here is coated with clinical-grade human serum albumin (HSA), and they have been substantially improved over their predecessors, and are readily scaled for the production of clinical quality reagents using current good manufacturing practices [cGMP]. The choice of injection grade HSA as the coating for FF was made because it is highly compatible with human cells, has negligible non-specific binding to them and also because very similar constructs have been used as MRI contrast agents with no toxic effects (G Doyle and D Gohel Immunicon Corp, personal communication). The version of HSA FF developed by us has HSA cross-linked on their surfaces by methods that do not apparently affect native structure. Further, no HSA leaches from these materials.
Here we report the development and optimization of a closed FF-based separation system in combination with an external-gradient planar magnetic separation device. For the development of this system, we chose the isolation of T cells from leukapheresis products because of its relevance and its degree of difficulty for testing this system. Non-target cell entrapment in IMCS is well known to be a function of the percentage of the population being separated, which ranges from 35–65% of total nucleated cells (TNC) for CD3+ cells from leukapheresis products. Accordingly T cell isolation is a stringent test of a separation system’s capability. We demonstrate that our external-gradient FF-enabled (X-GRAFFE) cell separation system is capable of producing T cells at high yield and high purity without an appreciable reduction in cell viability. These results are due to the gentle separation nature of FF in concert with externally generated gradients and the novel protocols that have been developed.
We intentionally designed our X-GRAFFE cell separation system to pair with existing cell processing systems. For this work we used a Fresenius Kabi’s Lovo Cell Processing System but other systems that provide similar functionality and performance could readily be used. When combined with our common-capture FF, planar magnetic array, blood-bag-based separation chamber, and in-field washing methodology Lovo possesses all the complementary components and capabilities necessary to create a closed, fully automated clinical-scale cell separator. By leveraging Lovo’s spinning membrane filtration technology, leukapheresis products can be depleted of platelets and plasma prior to antibody labeling, and excess unbound antibody can be removed following antibody labeling. In cases where ferrofluid cell labeling does not require mAb washout then the spinning membrane technology is not required. Lovo’s ability to easily concentrate and dilute cells allows for optimization of the antibody and FF labeling steps. Hence, leukapheresis products can be sterilely processed, target cells can be immunomagnetically labeled, and the cell product can be readily transferred sterilely to our X-GRAFFE cell separation system.
Materials & Methods
Materials
Because of the substantially high costs of aliquots of or full apheresis products, the strategy we used to develop a large scale cell separation system that would work in concert with a cell processing system was to do development at small scale system (2.0 to 10 ml), using quadrupole separators, with which each step of the protocol could perfected. Each improvement was next evaluated on X-GRAFFE with 100 ml aliquots of apheresis products, where removal of platelets, plasma and some RBC was done manually. Results from the small scale system translate quite well to X-GRAFFE. With each improvement of the protocol, a full apheresis product was done on X-GRAFFE using the Lovo cell processor. Components of the latter, including software, were used for platelet, plasma, some RBC reduction and also for uniformly mixing mAb, removal of unbound mAb, subsequent mixing of FF as well as volume reduction or increase where called for. Three such full apheresis products were done with X-GRAFFE in concert with the cell processor. The number of 100 ml aliquots that were processed on X-GRAFFE without the cell processor and between the full apheresis products done with Lovo was at least three. By doing development in this tandem manner with these three systems, we evolved to an optimal protocol and methodology for X-GRAFFE with and without automated cell processing. The results presented are a comparison of the optimized process performed on X-GRAFFE with and without a cell processor.
All references to ‘buffer’ refer to an iso-osmolar solution of modified Dulbecco’s phosphate-buffered saline (cat. no. 14200–075; 10X, free of calcium, magnesium, and phenol red) supplemented with 1% w/v bovine serum albumin (BSA; cat. no. 81–066) and 2 mM ethylenediaminetetraacetic acid disodium dihydrate (EDTA), adjusted to pH 7.8 with NaOH. All references to ‘sucrose-containing buffer’ refer to an iso-osmolar solution of 10 mM NaH2PO4 • H20, 70 mM NaCl, 2 mM EDTA, 5% w/v sucrose, and 1% w/v BSA, adjusted to pH 7.8 with NaOH. Human IgG (cat. no. 7403704; Protein A purified; 10 mg/ml in 0.01 M sodium phosphate, 0.15 M NaCl, pH 7.4) used for blocking prior to antibody labeling was purchased from Lampire Biological Laboratories (Pipersville PA). Biotinylated anti-human CD3+ (cat. no. 144–030; UCHT1 clone; 1 mg/ml in 50 mM sodium phosphate, 100 mM KCl, 150 mM NaCl, 5% glycerol, 0.2% BSA, 0.04% NaN3, pH 7.5) used for labeling of target cells was purchased from Ancell Corporation (Stillwater MN). Streptavidin-functionalized HSA-coated FF used for magnetically labeling antibody-labeled target cells was synthesized by BioMagnetic Solutions using a proprietary manufacturing process. Fluorescently tagged antibodies used for flow staining of cells recovered from X-GRAFFE of CD3+ cells (cat. no. 50–0039; Hit3a clone; labeled with PE) were obtained from Tonbo Biosciences (San Diego CA), and those used for flow staining of CD45+ cells (cat. no. 9625–02; F10–89–4 clone; labeled with FITC) and CD14+ cells (cat. no. 9560–13; clone UCHM-1; labeled with SPRD, a tandem dye comprising PE and Cy5) were obtained from Southern Biotech (Birmingham AL). Mouse IgG (cat. no. 7404304; Protein A purified; 10mg/ml in phosphate-buffered saline with 0.1% NaN3, pH 7.4) used for blocking prior to flow staining was purchased from Lampire Biological Laboratories (Pipersville PA).
Note that for the isolation of cells that would subsequently be used for therapy, buffers would use injection grade HSA instead of BSA to avoid the possibility of any undesirable Xeno reactions.
Design of the Planar Magnet Array
In preliminary small-scale experiments performed using 12×75 mm test tubes and quadrupole separators, it was observed that separated cells collect uniformly on the inside walls of the test tubes. It was discovered that non-target cells that were entrapped on the collection surface during separation could be effectively removed by adding and removing wash buffer while the tube was held in the quadrupole separator. It was hypothesized that the meniscus was able to force the non-target cells into the supernatant, thereby allowing them to be removed without the need to re-suspend and magnetically re-separate the cells. To examine the potential of this approach to be applied to large-scale separations, a planar magnet array was designed that would evenly collect target cells over as large an area as practicable.
To optimize the design of the planar magnet array, finite element method modeling employing Maxwell’s equations was carried out using freely available FEMM 4.2 software. Various magnet geometries and magnet spacings were modeled, and the magnetic field strength at various distances from the magnet array surface was investigated to provide insight into the ‘reach’ and ‘holding force’ for each modeled scenario. Additionally, the uniformity of the magnetic field gradient at the collection surface was investigated to provide insight into the ‘evenness’ of collection. The dimensions of the planar magnet array were chosen such that it could accommodate a large 2 l blood bag.
Design of the X-GRAFFE Cell Separation System
Because a sterile blood bag would provide a suitable chamber for separation and subsequent manipulations, experiments were initially performed wherein a blood bag was placed between a planar magnet array and a retaining plate such that when filled, the blood bag would form a quasi-rectilinear chamber with a height restricted to 5–10 mm. Although effective depletions of target cells could be achieved with such an arrangement, washing of magnetically collected cells for removal of entrapped non-target cells and quantitative recovery of target cells was difficult and inefficient due to the collapsible nature of the blood bag chamber. In addition to being difficult to create a flat, unwrinkled collection surface, the bag had a tendency to change shape and even collapse during filling, emptying, and wash manipulations; these issues led to the dislodgement of target cells and incomplete removal of non-target cells, resulting in lower yield, inadequate purity, and overall inconsistent results.
To create a rigid separation chamber with flat surfaces from a simple blood bag, we placed an appropriately designed flow-through bag (i.e., inlet and outlet on opposite sides of the bag) within a rigid-walled frame and inflated the bag via pneumatic pressure so that it formed a rigid quasi-rectilinear chamber wherein separation and other manipulations can take place. The walls of the frame which confine the bag are the magnet array on one side and a thick, transparent Plexiglas cover on the other side. This strategy of pressurizing the bag within a rigid frame allows separation to take place on a flat surface without the walls of the chamber falling away from the magnetic surface or collapsing during processing. The cost of this very high performing separation chamber that facilitates all required manipulations is insignificant and is readily manufactured to accommodate a range of volumes.
The way in which the magnetically labeled cells are collected on the magnetic collection surface and non-target cells are removed is also novel and confers numerous advantages. Because a large planar magnet array is used to evenly collect the magnetically labeled cells, target CD3+ cells from a typical leukapheresis product, for example, can be collected in only a couple layers of cells. This large magnetic collection surface allows non-target cells that are entrapped during magnetic separation to be readily removed by ‘in-field washing’– that is, removal of non-target cells while target cells are held in place magnetically. After collection, that is done by flushing the separation chamber with wash buffer to remove the majority of non-target cells, and then a volume of air is introduced to create an air space/meniscus. Next, the separation chamber–in tandem with the rigid frame including the magnet array–is rotated back and forth, causing the meniscus to pass over the magnetically collected cells to ‘scrub’ away non-target cells. In-field washing is effective because the surface tension of the meniscus pulls non-target cells off of the collection surface while FF-labeled target cells are held on magnetically. Compared to cycles of re-suspending and magnetically re-collecting cells, this gentle in-field washing strategy saves time; preserves target cell viability, and results in excellent yields and purities.
To achieve the foregoing, the planar magnet array is mounted at its center of gravity on a rotatable shaft, which is held on both sides of the planar magnet array by clamp assemblies to allow for 360°rotation of the array. A flexible bag is placed atop the planar magnetic array, after which a 3/8 thick Plexiglas cover piece is mounted onto the planar magnet array’s backing plate at a variable distance above the planar magnet array (nominally a 6 mm spacing). Upon inflation of the bag, a rigid separation chamber of appropriate volume is formed. A second flexible bag is connected to the outlet of the separation bag, and this bag is also inflated and fitted with a pressure relief valve. While this bag primarily serves as a waste reservoir, a secondary purpose is to act as a bladder. All solutions are pumped into and out of the bag using a peristaltic pump. Figure 1
Choice of the Cell Labeling Method
Two cell labeling methods exist for IMCS: direct labeling, wherein cells are incubated with magnetic particles functionalized with monoclonal antibody (mAb), and indirect labeling, wherein cells are first incubated with mAb, followed by removal of unbound mAb and subsequent addition of common-capture magnetic particles (e.g., streptavidin-functionalized particles); the direct protocol is typically employed by Miltenyi. From thorough preliminary work, we have determined that the indirect labeling approach is significantly superior to the direct labeling approach in terms of yield and purity (unpublished results). Based on those findings, along with the significant cost savings and versatility it affords, an indirect labeling approach with a clinical-grade common-capture FF (i.e., streptavidin-FF) and biotinylated mAb was used in the development of our X-GRAFFE cell separation system.
Isolation of CD3+ Cells from Leukapheresis Product with Manual Platelet/Plasma Removal
Leukapheresis products were obtained from Key Biologics (Memphis TN), and were collected from a normal, healthy donor using a Fenwal Amicus system (Fresenius Kabi Lake Zurich IL). On the same day that they were collected, leukapheresis products were shipped overnight with a room temperature gel pack and were used immediately upon arrival at our facility. An in-process bag (referred to as the separation bag henceforth) was taken from a Lovo single-use disposable cell processing set (XR4909, Fresenius Kabi), and its top offset port was connected to a 2 l transfer bag (referred to as the waste bag henceforth), which served to collect non-magnetically labeled cells during separation. A secondary port on the waste bag was connected to a regulator through a liquid trap, permitting pressurization of the waste bag and providing pressure relief as the waste bag was filled. Prior to beginning the experiment, lines and bags were primed as necessary, and the waste bag was pressurized to approximately 1 psi with the line between the waste and separation bags clamped to prevent premature pressurization of the separation bag.
The leukapheresis product was initially depleted of platelets and plasma using repeated low-speed centrifugation to prepare platelet-rich plasma. The leukapheresis product was placed into 50 ml conical centrifuge tubes and centrifuged at 250 g for 10 min, after which the supernatant was decanted. This process was repeated five times, and after the final supernatant removal, cells were re-suspended in a volume of buffer to yield a final total nucleated cell (TNC) concentration of 2.5 × 107TNC/ml. A volume of human IgG was added to the cell suspension to yield a final IgG concentration of 1mg/ml and a final cell concentration of 2.2 × 107 TNC/ml, and following a brief agitation, was statically incubated for 10 min. Subsequently, a volume of biotinylated anti-human CD3 in buffer was added to the cell suspension to yield a final mAb concentration of 1.5µg/ml and a final cell concentration of 2 × 107 TNC/ml, after which the cell suspension was briefly agitated and statically incubated for 10 min. Upon completion of the mAb incubation, two cycles of centrifugation (250 g for 10 min) and supernatant removal were used to deplete the cell suspension of excess unbound mAb, and after the final supernatant removal, cells were re-suspended in a volume of buffer to yield a final cell concentration of 4 × 107 TNC/ml. A volume of FF in buffer was added to the cell suspension to yield a final FF concentration of 8g/ml and a final cell concentration of 2 × 107 TNC/ml, after which the cell suspension was briefly agitated and statically incubated for 15 min.
Following magnetic labeling, the cell suspension containing magnetically labeled target cells and unlabeled non-target cells was diluted to a concentration of approximately 1.33 × 107 TNC/ml with buffer. The separation bag was placed into its rigid frame, pressurized by opening the clamp between the separation bag and the inflated waste bag, and a known volume of the cell suspension was pumped into the separation bag via a peristaltic pump (cat. no. 146821, Spectrum Laboratories Rancho Dominguez CA). Once the entire cell suspension was pumped into the separation bag, it was allowed to separate for 15 min. Following separation, the separation assembly (i.e., the rigid frame containing the pressurized separation bag) was tilted to approximately 45 such that the inlet of the separation bag was lower than the outlet, and approximately 250 ml of sucrose-containing buffer was pumped into the rigid bag separation chamber to force the negative fraction to waste. Following removal of the negative fraction, a volume of air was pumped into that chamber to form a meniscus, and the separation assembly was manually rocked back and forth 20 times to perform a meniscus scrub. Subsequently, a volume of sucrose-containing buffer was pumped into the separation bag with the separation assembly tilted to approximately 45 to force the air out of the separation bag, and a 5 min waiting period was observed before proceeding to the next step to allow any target cells that might have been dislodged during the meniscus scrub to re-collect on the magnetic surface. Subsequently, buffer (ca. 250 ml) was pumped into the separation bag with the separation assembly tilted to approximately -45 such that the outlet of the separation bag was lower than the inlet. Air was introduced, a second meniscus scrub was performed, and the air was removed by pumping buffer into the separation bag with the separation assembly at 45. After 5 min, sucrose-containing buffer (ca. 250 ml) was pumped into the separation bag with the separation assembly tilted to approximately 45. Air was introduced, a third meniscus scrub was performed, and the air was removed by pumping sucrose-containing buffer into the separation bag with the separation assembly at 45. After 5 min, buffer (ca. 250 ml) was pumped into the separation bag with the separation assembly tilted to approximately -45, and the separation bag was subsequently removed from the rigid frame–and thus, the magnetic field–to recover the purified target cells. Samples were taken from the original leukapheresis product, the final purified product, and throughout the process to measure the target and non-target cell concentrations as well as cell viability; these data were used to characterize the process in terms of yield, purity, and viability.
Use of Lovo Cell Processing System in the Isolation of CD3+ Cells from Leukapheresis Product
Leukapheresis products were collected from normal, healthy donors in an on-site collection facility using a Fenwal Amicus system (Fresenius Kabi Lake Zurich IL) and used immediately following collection. A Lovo single-use cell processing set (XR4909, Fresenius Kabi, shown in Figure 2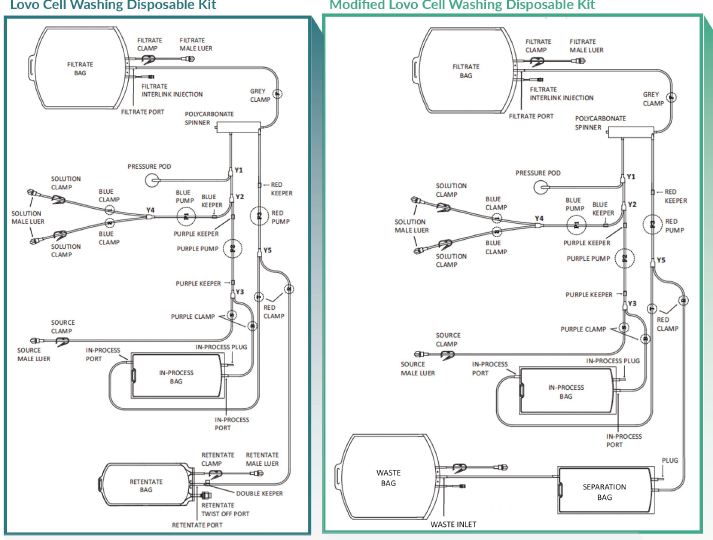
The leukapheresis product was initially depleted of platelets and plasma using an automated two-cycle wash with buffer. At the end of the two-cycle wash, cells were concentrated to a target volume of 130 ml in the in-process bag. Human IgG (15 ml) in buffer was added to the in-process bag through an injection port to yield a final IgG concentration of 1 mg/ml, after which the cell suspension was manually agitated and statically incubated for 10 min. Subsequently, biotinylated anti-human CD3 (15 ml) in buffer was added to the in-process bag through an injection port to yield a final mAb concentration of 1.5µg/ml, after which the cell suspension was manually agitated and statically incubated for 10 min. Upon completion of the mAb incubation, a two-cycle wash was used to deplete the cell suspension of excess unbound mAb, with cells concentrated to a target volume of 100 ml in the in-process bag at the end of the wash. FF (60 ml) in buffer was added to the in-process bag through an injection port to yield a final FF concentration of 8g/ml, after which the cell suspension was manually agitated and statically incubated for 15 min.
Following magnetic labeling, the entire 160 ml cell suspension containing magnetically labeled target cells and unlabeled non-target cells was transferred to the separation bag and approximately 30 ml of buffer was used to rinse the lines. After weighing the separation bag to estimate the volume of cell suspension contained within, the separation bag was placed into its rigid frame, pressurized by opening the clamp between the separation bag and the inflated waste bag, and allowed to separate for 15 min. Following separation, the identical steps that were used for the manual platelet/plasma removal experiment, above, were followed except the third meniscus scrub was done with about 275 ml of buffer. Also samples were taken from the original leukapheresis product, the final purified product, and throughout the process as above.
Determination of Cell Concentration and Cell Viability
In the experiment where platelets and plasma were removed manually, cell concentration and cell viability determinations were made using a Cellometer Auto 2000 Cell Viability Counter (Nexcelom Lawrence MA); the same instrument and technique was used for cell viability determinations only in the experiment where the Lovo Cell Processing System was employed. A 15 µL sample of cell suspension was mixed with 15 µL of a solution containing the nuclear stains acridine orange and propidium iodide, and 20 µL of the mixture was loaded into a Cellometer Disposable Imaging Chamber (CHT4-SD100–002, Nexcelom Lawrence MA). Acridine orange stains both live and dead nucleated cells, while propidium iodide only stains dead nucleated cells (non-nucleated cells such as erythrocytes are not stained). Due to quenching of the acridine orange’s green fluorescence in doubly stained dead cells, only red fluorescence from propidium iodide is visible. Consequently, live cell concentrations were determined based on counts of green fluorescent cells, while cell viability was garnered from the ratio of green fluorescent cells to total fluorescent cells (i.e., green + red fluorescent cells).
Determination of Cell/Platelet Concentrations for Experiment Employing the Lovo Cell Processing System
A hematology analyzer (KX-21N, Sysmex Kobe Japan) was used to determine the concentrations of TNC, erythrocytes, and platelets in the source leukapheresis product, the purified product, and intermediate samples. Operating in whole blood mode, a 50 µL sample is required to provide accurate concentration determinations. The linear ranges are 1.0–99.9 × 103 TNC per µL, 0.30–7.00 × 106 erythrocytes per µL, and 10–999 × 103 platelets per µL.
Flow cytometry
To determine the relative proportions of the various cell types at different stages of the purification process, flow cytometry was employed. Samples taken for flow cytometric analysis were first concentrated or diluted to 1–2 × 107 TNC/ml (where necessary) and blocked for 10 min with 0.2mg/ml mouse IgG. Subsequently, 5–10L (per the manufacturer’s recommendations) of the fluorescently tagged antibodies were added to each sample and allowed to incubate on ice for 20 min (singly stained samples were prepared as well for construction of a compensation matrix). Unbound fluorescently tagged antibody, as well as mouse IgG and BSA, were removed via two centrifugation cycles and the sample was fixed with 1% paraformaldehyde for 20 min (fixing was not performed for the experiment where the Lovo Cell Processing System was employed).
For the experiment where platelets and plasma were removed manually, an Amnis FlowSight imaging flow cytometer (Millipore Sigma Burlington MA) was used to collect the data, and IDEAS software (Millipore Sigma Burlington MA) was used for data analysis. For the experiment where the Lovo Cell Processing System was employed, a FACS Canto II system (BD Biosciences Franklin Lakes NJ) was used to collect the data, and FACSDiva software (BD Biosciences Franklin Lakes NJ) was used for data analysis. A compensation matrix was constructed using the three singly stained samples, after which the samples were loaded. A first gate was used to exclude platelets and debris (area vs aspect ratio for the FlowSight; side-scatter vs forward-scatter for the FACS Canto II), and a second gate was used to exclude CD45- cells. From the CD45+ cells, gates were placed to identify CD3+ cells and CD14+ cells.
Results
Design of the Planar Magnet Array
Through modeling, we determined that 200 mm long x 5 mm wide x 20 mm tall block magnets magnetized through their height, spaced 3–4mm apart, and arrayed across a slotted iron backing plate perpendicular to the direction of flow in and out of the bag provide an optimal magnetic field gradient which collects cells quite uniformly across the planar surface of the magnet array. The dimensions of the magnet array easily accommodate the separation bag that we employed in these experiments, but could accommodate a larger separation bag as well to process larger blood products (e.g., mobilized blood products for CD34+ cell isolation) if desired. Similarly, by employing a smaller separation bag, smaller blood products (e.g., cord blood) could be processed as well, offering valuable user flexibility.
Isolation of CD3+ Cells from Leukapheresis Product with Manual Platelet/Plasma Removal
A 113 ml leukapheresis product (3.8% hematocrit [hct]) was received and processed as described in the Materials and Methods section. After depleting platelets and plasma, 81 ml of buffered cell suspension containing 2 × 109 TNC was evenly divided between two 50 ml centrifuge tubes, and 4.5 ml of human IgG was added to each tube. After 10 min, 5 ml of biotinylated anti-human CD3 was added to each tube and allowed to incubate for 10 min. Following two centrifugation cycles to deplete the blocking agent and unbound mAb, cells (1.84 × 109 TNC, 8% loss during centrifugation) were re-suspended in 46 ml of buffer, to which 46 ml of FF was added and incubated for 15 min. Buffer (46 ml) was then added to dilute the cell suspension for separation.
Prior to separation, samples (n = 2) were taken for measurements of cell concentration, viability determination, and analysis by flow cytometry. The concentration of TNC in the labeled cell suspension (135.75 ml) prior to separation was determined to be 136 ± 2.36 × 105 TNC/ml (184 ± 3.21 × 107 TNC total), and the viability of cells in this starting preparation was 91.95 ± 1.58%. Flow cytometric analysis revealed that the pre-separation sample was 69.6 ± 0.42% CD3+, 9.82 ± 1.39% CD14++, and 1.55 ± 0.20% CD14+.
Following separation, 247 ml of purified cell suspension was recovered from the separation bag (Fraction 1), approximately 60 ml of buffer was pumped into the separation bag for rinsing, and 59 ml of diluted purified cell suspension was recovered (Fraction 2). Samples from Fraction 1 (n = 4) and Fraction 2 (n = 2) were taken for measurements of cell concentration, viability determination, and analysis by flow cytometry. The concentration of TNC in Fraction 1 was determined to be 360 ± 9.98 × 104TNC/ml (88.8 ± 2.47 × 107 TNC total), and the viability was 92.90 ± 0.96%. The concentration of TNC in Fraction 2 was determined to be 23.8 ± 3.18 × 104 TNC/ml (14.0 ± 1.88 × 106 TNC total), and the viability was 89.00 ± 8.77%. Given that the two fractions collectively contained 9.02 × 108 TNC, the process yield was approximately 49% (i.e., TNC recovered from total input TNC). Flow cytometric analysis revealed that the purified cell suspension was 93.1 ± 0.28% CD45+, and of the CD45+ cells, 97.9 ± 0.00% were CD3+, 0.72 ± 0.15% were CD14++, and 0.31 ± 0.00% were CD14+. Therefore, the yield of CD3+ cells was approximately 69%.
Use of a Cell Processing System in the Isolation of CD3+ Cells from Leukapheresis Product
A 238 ml leukapheresis product (1.9% hct) was collected and found to contain 11.25 × 106 TNC/ml (99.9% viability), 0.20 × 109 erythrocytes/ml, and 213 × 106 platelets/ml. Of the TNC, 58.1% were CD3+ and 11.9% were CD14+. Following a two-cycle wash to deplete platelets and plasma, 97.3% of platelets and 35.1% of erythrocytes were removed, while 95.1% of the TNC were retained and their viability was 97.7%. Following mAb incubation and a second two-cycle wash, the platelet concentration was below the detection limit and the erythrocyte level was further reduced to 51.7% of its initial value. After FF incubation, the cell suspension was diluted with buffer and transferred to the separator.
Prior to separation, samples were taken for measurements of cell concentration, viability determination, and analysis by flow cytometry. The concentration of TNC in the labeled cell suspension (209 ml) prior to separation was determined to be 13.1 × 106 TNC/ml (2.74 × 109 TNC total), and the viability was 95.8%. Flow cytometric analysis revealed that the pre-separation sample was 62.1% CD3+ and 11.2% CD14+.
Following separation, 275 ml of purified cell suspension was recovered from the separation bag. Samples were taken for measurements of cell concentration, viability determination, and analysis by flow cytometry. The concentration of TNC in the purified cell suspension was determined to be 6.2 × 106 TNC/ml (1.71 × 109 TNC total), and the viability was 94.0%. Taking into account the losses due to sampling, the process yield was 65.1% (i.e., TNC recovered from total input TNC). Flow cytometric analysis revealed that the purified cell suspension was 99.9% CD45+, and of the CD45+ cells, 92.8% were CD3+ and 5.0% were CD14+. Therefore, the yield of CD3+ cells by this process was 93.1%.
Discussion
Employing external magnetic gradients for separations confers significant advantages not only because desirable collection patterns can be achieved by optimally shaping gradients, but also because separations can be done in simple and reasonably inexpensive vessels. The need to create a separation chamber that can:
- Maintain sterility throughout the purification process;
- Be part of a sterile closed system
- Collect target cells over a large area in order to minimize entrapment of non-target cells; and
- Separate target cells in such a manner that the removal of entrapped cells can be accomplished with minimal manipulation, indeed, presents a number of challenges
Additionally, there are a number of boundary conditions to be considered, such as FF-labeled cells–even with very strong external magnetic gradients–can only be efficiently separated in a reasonable time period (i.e., We found that such an arrangement allows a) pumping a cell mixture containing FF-labeled cells into the bag-based separation chamber; b) magnetic separation; c) pumping wash buffer through the separation chamber to remove unseparated cells; and d) subsequent cycles of passing wash buffer with controlled volumes of air over the magnetically held cells to remove entrapped non-target cells. Even though this process of removing non-target cells while magnetically holding target cells in place on the bag’s collection surface showed promise, the necessity for a flat collection surface and a rigid container became apparent as the blood bag would only be held against the walls confining it (i.e., the planar magnet array and the retaining plate) when the bag was completely filled; the bag would partially or completely collapse during subsequent processing steps. To address this issue, we improvised an arrangement whereby the blood bag was pressurized such that it would be confined to a rectangular space between the planar magnet array and a cover piece. Thus, by maintaining the blood-bag-based separation chamber under a constant positive pressure throughout the entire purification process, a flat, non-collapsible collection surface is achieved and a rigid chamber in which multiple operations can be performed is created. We are currently in the process of engineering into X-GRAFFE synchronous pumps on the inlet and outlet ends of the separation chamber that will maintain rigidity of the chamber by hydraulic pressure and a combination of hydraulic and pneumatic pressures for steps such as ‘in-field’ washing.
This innovative rigid separation chamber formed from a flexible blood bag–along with the gentle magnetic separation of FF-labeled cells and the ability to remove non-target cells while maintaining target cells on the collection surface–allows one to obtain high yields, purities, and viabilities of target T cells using the X-GRAFFE cell separation system. The process of washing away non-target cells was made remarkably efficient by introducing wash buffer along with a controlled bolus of air into the separation chamber. The purpose of the air bubble is to create a meniscus that moves over the collection surface of the separation chamber as it is gently rocked back and forth. This gentle meniscus scrubbing effect utilizes surface tension to create enough force to pull non-target cells off of the collection surface.
The depth of the separation chamber was designed to be variable (i.e., 5–15 mm) to test collection efficiency as a function of depth, as well as to provide the ability to accommodate large sample volumes (i.e., up to ca. 380 ml). Another advantage of a variable-depth chamber is to facilitate optimization of the various processing steps. For example, a depth of 10–15 mm is useful to maximize the volume to be separated, but for subsequent steps, such as buffer wash of magnetically held target cells, lesser depths might be desirable to preserve wash buffer and save time. Aside from being able to control the volume of the separation chamber by altering the chamber depth, the X-GRAFFE cell separation system can accommodate bags of smaller dimensions such as what might be required for cord blood samples. On the other hand, there is no reason that extremely large samples cannot be processed via multiple fillings of the separation chamber.
Another feature of the X-GRAFFE cell separation system is that because the rigid frame containing the separation chamber and comprising the planar magnet array is mounted on a rotatable shaft, the entire device can be rotated 360o, permitting magnetic separations to be performed either with or against gravity. Although all the separations reported here for the X-GRAFFE cell separation system are performed with gravity (i.e., separation bag on top of planar magnet array), we have demonstrated feasibility that separations can indeed be performed against gravity, and preliminary results show a reduction in non-target cells entrapped in the initial magnetic separations. In fact, erythrocytes can be visually observed falling downwards from the collection surface when target cells are separated from leukapheresis products against gravity. As a consequence, fewer cycles of in-field washing were required to obtain highly purified target cells. Thus incorporation of separations against gravity to the X-GRAFFE cell separation system should shorten the processing time.
We have demonstrated in previous work [19] that T cells isolated from PBMC using a rat anti-mouse IgG1-conjugated common-capture FF in conjunction with mouse-derived human anti-CD3 mAb (IgG1 subclass) could be activated as determined by their expression of activation markers, (i.e., CD69 and CD25). When such purified, activated T cells were cultured in IL-2-containing expansion media and stimulated with solution-phase anti-CD28 mAb, expansions as high as 600-fold in 18 days were observed, indicating that the isolated cells are functional. The demonstration that 3 days after activation, cells can be transfected with a green fluorescent protein plasmid with high viability and efficiency is further evidence of functionality. Thus the X-GRAFFE cell separation system, when used with appropriate reagents and protocols, is capable of simultaneously separating and activating T cells.
Compared with other commercially available systems that have been used, the X-GRAFFE cell separation system we have described here has significant advantages. In performance, the X-GRAFFE cell separation system provides purities comparable to Miltenyi’s systems and the Isolex in its various iterations. When using the Lovo Cell Processing System, the purity of CD3+ cells as a fraction of CD45+ cells was lower (92.8% vs 97.9%); however, when accounting for the purity of CD45+ cells (99.9% vs 93.1%), the overall purity of CD3+ cells (92.7% vs 91.1%) was comparable to manually depleting leukapheresis products of platelets and plasma. One explanation for this could be that Lovo reduced the number of erythrocytes by approximately half, and because the hematocrit level for that leukapheresis sample was already half (1.9% htc vs 3.8% htc), the overall number of erythrocytes in the initial separation was approximately 25%. As regards yields, the X-GRAFFE cell separation system is significantly superior, and in some cases, nearly two times better. Using Lovo resulted in a drastic increase in the CD3+ cell yield (93.1% vs 69%), which could be explained by Lovo’s superior ability to remove platelets and erythrocytes (sources of mAb and FF non-specific binding) coupled with a significantly faster processing time and the introduction of in-field meniscus scrubbing. The yield superiority of X-GRAFFE compared with other systems suggests smaller apheresis products may suffice.
Because of the gentle nature of the processing steps, as well as the short processing time ( As regards the purities of CD3+ cells isolated by X-GRAFFE, although acceptable and comparable to other large scale systems, we are confident they can be improved merely by improving the design of the flow thru blood bag used to create the separation/process chamber for these experiments. The bags employed are of large rectangular shape with entry and exit ports placed at the centers of the short sides. From simple experiments of just filling and emptying such bags with dilute RBC suspensions, made into a rigid chamber as described here, cells clearly are not efficiently flushed out simply because they get trapped in corners. A rectangular bag design with tapered ends should resolve that issue and lead to higher purities. For separations done in test tubes and quadrupole separators which mimic the X-GRAFFE process, purities of 98.0–99.9 % are routinely obtained.
The marriage of X-GRAFFE incorporating an indirect magnetic cell labeling approach and a wide range of process volume choices with a cell processor such as a Fresenius Lovo cell processing system is clearly beneficial in terms of function, versatility as well as economy in development cost and costs to the end user. We are exploring X-GRAFFE’s use with cell elutriation processors such as those of Terumo that could provide a better starting population of cells with which to work.
The combination of the benefits of X-GRAFFE with a commercial cell processor is best illustrated with a flow diagram (Figure 3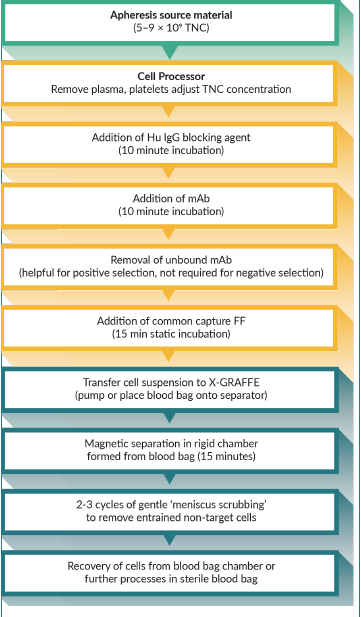
Conclusions
By pairing our X-GRAFFE cell separation system with a cell Processing System and employing new simplified techniques for performing immunomagnetic cell separations that reduce labeling times, an entire process starting with leukapheresis product and ending with purified CD3+ cells can be completed in a few hours. By utilizing a novel high-gradient-producing planar magnet array for retrieval of FF-labeled target cells within an appropriately designed blood bag to distribute collected cells over a large area, target cells can be recovered without the need for suspension and re-separation to free entrapped non-target cells, resulting in better yields, higher purity, and improved viability of the product. In summary, we have developed an automatable system with built-in versatility for clinical-scale cell separation that is faster, cheaper, and better performing (in terms of yield, purity, and viability) than currently available cell separation systems, defining X-GRAFFE as the new state of the art.
Acknowledgements
The authors gratefully acknowledge Augustin Min and Christopher Wegener and their colleagues at Fresenius Kabi Lake Zurich IL for supplying leukapheresis products, as well as making available the use of their hematology analyzer, flow cytometer, and a Lovo Cell Processing Unit. The Microscopy and Cytometry Facility in The Pennsylvania State University’s Huck Institutes of the Life Sciences is acknowledged for use of their imaging flow cytometer. We are also grateful to Ms. Sai Patkar for her excellent technical assistance and to Dr. Qiuyan Chen for her insightful comments.
This work was supported in part from an SBIR Grant No. 1R43HL131402–01 from the NHLBI of the NIH: A RAPID, HIGH PERFORMANCE, COST-EFFICIENT CLINICAL SCALE SEPARATOR FOR CD3+ CELLS .
References
1. Molday RS, Yen SPS, Rembaum A. Application of magnetic miscrospheres in labeling and separation of cells. Nature 1977; 268, 437–438.CrossRef
2. Molday RS, Molday LL. Separation of cells labeled with immunospecific iron dextran microspheres using high gradient magnetic chromatography. FEBS Letters 1984; 170(2), 232–238.CrossRef
3. Owen CS. High gradient magnetic separation of erythrocytes. Biophysical J. 1978; 22(2), 171–178.CrossRef
4. Kemshead JT, Ugelstad J. Magnetic separation techniques: their application to medicine. Molecular and Cellular Biochemistry 1985; 67(1), 11–18.
5. Miltenyi S, Müller W, Weichel W, Radbruch A. High gradient magnetic cell separation with MACS. Cytometry 1990; 11(2), 231–238.CrossRef
6. Zborowski M., Commercial magnetic cell separation instruments and reagents. In Laboratory Techniques in Biochemistry and Molecular Biology Elsevier: 2007; Vol. 32, 265–292.CrossRef
7. Liberti PA, Feeley BP, Gohel DI. Apparatus and methods for magnetic eparation. US 5200084 A, Sept 26, 1990, 1993.
8. Liberti PA, Rao GC, Chiarappa JN. Methods for the manufacture of magnetically responsive particles. US 5698271 A, Jun 7, 1995, 1997.
9. Liberti PA, Rao GC, Chiarappa JN. Coated, resuspendable magnetically responsive, transition metal oxide particles and method for the preparation thereof. US 6120856 A, Filed:Oct 14, 1997, Published 2000.
10. Liberti PA, Rao GC, Terstappen LWMM. Increased separation efficiency via controlled aggregation of magnetic nanoparticles. US 6623982 B1, Filed: July 12, 1999, Published: 2003.
11. Liberti PA, Wang Y. Methods and devices for manipulation of magnetically collected material. US 5622831 A, Filed: Jun 7, 1995, Published: 1997.
12. Liberti PA, Wang Y. Magnetic immobilization and manipulation of biological entities. US 5876593 A, Filed: Sep 15, 1997, Published: 1999.
13. Liberti PA, Wang Y, Tang W, Feeley BP, Gohel DI. Apparatus and methods for magnetic separation featuring external magnetic means. US 5466574 A, Filed: Jan 15, 1993, Published: 1995.
14. Rao GC, Terstappen L, Liberti P. Methods for enhancing binding interactions between members of specific binding pairs. US 6551843 B1,Filed: Jan 29, 1999, Published: 2003.
15. Terstappen LWMM, Rao GC, Uhr JW, Racila EV, Liberti PA. Methods and reagents for the rapid and efficient isolation of circulating cancer cells. US 6645731 B2, Filed: July 13, 2001, Published: 2003.
16. Terstappen LWMM, Rao GC, Uhr JW, Racila EV, Liberti PA. Methods and reagents for the rapid and efficient isolation of circulating cancer cells. US 7332288 B2, Filed: Oct 11, 2002, Published: 2008.
17. de Haën C. Conception of the First Magnetic Resonance Imaging Contrast Agents: A Brief History. Topics in Magnetic Resonance Imaging 2001; 12(4).CrossRef
18. Ito A; Shinkai M; Honda H; Kobayashi T. Review: Medical application of functionalized magnetic nanoparticles Journal of Bioscience and Bioengineering, 2005; 100, 1–11.CrossRef
19. Ritter DW, Khristov TR, Liberti PA. Abstract 1612: Production of CD3+ cells using ferrofluids for cell isolation, activation, expansion and subsequent transfection for adoptive cell therapy. Cancer Research 2017; 77(13 Supplement), 1612–1612.CrossRef
Affiliations
Corresponding author: PA Liberti, BioMagnetic Solutions LLC, State College PA 16803, USA PLiberti@BioMagneticSolutions.com
DW Ritter, BioMagnetic Solutions LLC, State College PA 16803, USA
TR Khristov, BioMagnetic Solutions LLC, State College PA 16803, USA