Strategic raw material selection for cell therapy commercialization
Cell & Gene Therapy Insights 2024; 10(1), 19–28
DOI: 10.18609/cgti.2024.26
In the ever-evolving global landscape of cell therapy development, selecting quality raw materials is crucial for achieving clinical and commercial milestones. In this Innovator Insight article, Kyle Hondorp asks industry experts, Kasey Kime and Lili Belcastro, to share insights into the key factors influencing raw material selection throughout different phases of development. Kyle Hondorp also discusses the Gibco™ Cell Therapy Systems (CTS™) portfolio of fit-for-purpose media and reagents, cell therapy instrumentation, and viral vector systems, which are GMP manufactured, safety tested, and backed by regulatory documentation to support cell and gene therapy developers along their path to commercialization.
KH: What are key factors that organizations should consider when selecting critical raw materials to cater to both early- and late-phase development requirements? How can this help ensure a seamless transition during development?
KK: It is important to plan for success, and this involves discussing the regulatory documentation early in the development process. My advice is to review your supplier documentation from both a raw material criticality and a phase-appropriate perspective. This is going to help you identify if you think that there will be a need for any additional information about your raw material.
If you think there might be a need for any additional information, have these discussions early and understand what your supplier can offer you. For example, some suppliers can offer raw material master files. This can be a very efficient regulatory pathway for both the drug developer and the supplier. However, some suppliers do not have master files and some regulatory jurisdictions do not accept them. In those cases, you must have an early discussion with your supplier about how they can support you. They might offer you some kind of regulatory documentation package under a confidentiality agreement.
LB: One key factor when selecting critical raw materials is thinking about the ‘grade’ of the materials. Is it manufactured under GMP conditions? Is it being marketed as a research use-only (RUO) product? Knowing this information makes it easier to move from early-phase into late-phase development. If you start with the highest quality, GMP manufactured materials, you decrease the need for comparability studies in late-phase development. However, non-GMP or RUO materials are usually cheaper, so some companies may want to use them in early development to help save money.
Additionally, as you move from early into late phases, it is important to begin thinking about sourcing. Having a dual source for your raw materials is beneficial, but it is important to introduce the dual sources into your process as early as possible. This will avoid the need to do comparability work later on. When using a sole source for your material, you should ensure that the manufacturer has redundant manufacturing capabilities.
KH: How can organizations strike the right balance when selecting raw materials to optimize their production processes while managing costs?
LB: As I mentioned previously, there are options for using cheaper materials, which can help reduce costs in early-phase development. In fact, there are no firm requirements that only GMP materials can be used in commercial manufacturing, but that said, using GMP materials is definitely a best practice to adhere to—if you do not use them, health authorities will ask why you chose a lower-quality material when a higher-quality material was available.
The United States Pharmacopeia (USP) 1043 chapter provides guidelines on materials that are very well characterized and have, for example, a national drug code associated with them, as opposed to materials that are RUO, for example. As you move from a higher quality material to a lower quality material, as per USP 1043, there is going to be an increased amount of testing, documentation, and lot-to-lot comparability assessment of the incoming material. It is therefore up to the user to determine whether starting with GMP materials throughout the process might be more cost-effective than using a lower-quality material and having to perform additional testing.
KK: To reiterate, you want to make sure in early-phase development that you have done your due diligence on your raw materials, particularly with regard to safety risks like viral safety or sterility. Doing this exercise early is going to help set you up for filing success later on in the process.
KH: Kasey, you mentioned screening raw materials early and assessing whether any additional information is needed. Can you share some examples of additional documentation that might be required?
KK: One good example is details on adventitious viral agent testing for human or animal origin materials, or further information around viral inactivation and viral clearance. Often, if a supplier has a master file, these details will be in there, so you would not have to worry about them. But for regions that do not accept master files or for suppliers that may not have master files, these are the instances where you want to have those early discussions about the raw material to make sure that you are set up later for any regulatory questions that you anticipate.
KH: Could you comment on which regions do allow for raw material master files?
KK: The USA, Canada and Japan all allow for raw material master files. Sometimes, you can submit directly to a health authority on a sponsor’s behalf but this approach is not always accepted. We do know that Europe is undergoing review of their pharmaceutical regulations and there is a push to enable raw material master files in Europe. This would be a very welcome amendment.
KH: It sounds like having discussions early on with your suppliers can really help ensure you are aligned on the types of regulatory documentation they are able to support as well as provide awareness of your supplier’s continuity plans prior to reaching late-phase clinical trials.
Moving on to the growing demand for rapid drug development in cell therapy, what are the unique challenges that the industry faces in selecting and procuring raw materials, and how can these challenges be addressed effectively?
KK: Many cell and gene therapies are eligible for expedited drug development, meaning that they move quickly through the clinical trial phases. It is important to consider whether your supplier can actually scale to meet your commercial demand. For example, at Thermo Fisher, we have seen requests to scale-up five times in as little as 3 years for some cell culture media products.
You should also consider change management. Pre-notification of changes to critical raw materials used in commercial processes is important to manage your post-approval CMC obligations. Occasionally at Thermo Fisher, we observe instances where late-phase customers have not subscribed to change notifications, which can cause them unnecessary challenges such as an unexpected comparability study if a change is made to a product.
LB: The landscape for cell and gene therapy drug products has been dramatically changing over the past few years. The same goes for the raw materials that are used in cell and gene therapy manufacturing. Just a few years ago, many of the materials used in the cell therapy manufacturing process, for example, were sole-source materials because there were no other options available on the market.
With the rapid growth in the number of specialist companies providing additional options for different raw materials, it is important to perform technical due diligence for each vendor. It is also important to assess the overall raw material risk before you go about selecting a new material or a new supplier. The challenge here is that there are some vendors who are relatively new to cell and gene therapy drug product manufacturing, so they may not be familiar with the specific raw material requirements in this space. It is the responsibility of the user of these raw materials to ensure that this potential second source provider is not only providing good quality, robust materials, but that they are also a robust supplier in terms of their quality systems and their GMP manufacturing processes.
KH: What are some of the main considerations when evaluating a raw material supplier? What criteria should organizations prioritize when choosing suppliers to meet their specific needs?
LB: When you are evaluating a new supplier, you must look at the quality of the material, assess the material in your process, and retest any critical attributes that might be on the certificates of analysis (CoA). Before you get to that point though, you need to evaluate the supplier itself. For example, you must evaluate the supplier’s top criteria, the GMP manufacturing processes in place, and the quality management systems that are employed. Does the company have a business continuity or a disaster recovery plan B? Do they have regulatory experience? Have they helped support in the filing of any cell and gene therapy drug products before? Do they have a drug master file or a regulatory support file? What is their general experience in the industry?
KK: Again, I would focus on documentation. Having access to adequately detailed and specific CoA is important because these are key documents needed for regulatory filing. Currently, there is no standardization for the way in which raw material quality attributes are reported on the CoA. This can lead to variability amongst suppliers. However, there is some general guidance provided within ISO 20399, and this is an area of focus for some industry groups such as the Standards Coordinating Body.
KH: How could organizations ensure the quality and consistency of raw materials throughout the different phases of development and production? Are there any best practices or standards that can be followed?
LB: Materials can be handled in a phase-appropriate manner for cell and gene therapy manufacturing. For example, in early-phase development, verification of the CoA can be an acceptable incoming specification for raw materials. As projects move into late-phase development and commercialization, you should have some testing specification with critical quality attributes that are important for your final product and your process. At the bare minimum, ensure that the methods are validated, and that identity and safety testing is done.
Luckily, there are a lot of really great best practices and standards that are now available, like the USP 1043 chapter for ancillary materials and the ISO 20399 standard for ancillary materials. In addition, there is a lot of great documentation in BioPhorum, including a raw material risk assessment that can help with determining the best materials and the best practices. There are also some ongoing efforts to standardize raw materials with a standardized CoA.
KK: I agree with Lily. The various pharmacopoeias’ general chapters on raw materials used in cell and gene therapy manufacturing provide excellent advice on raw material selection and qualification. There are also some product-specific US FDA CMC guidelines. For example, ICH Q8 and Q9 provide details of how to implement QbD and quality risk management into raw material selection.
KH: In an era of growing environmental and ethical concerns, how can raw material selection align with sustainability and ethical sourcing practices? Are there any emerging standards in this regard?
KK: This is an increasingly important topic, especially considering the efforts to reduce animal testing in certain raw material testing procedures. A good example of this is USP 88, in vivo biological reactivity testing, which is an animal-based test for single-use systems that are used in further manufacturing applications. There is a push from industry to discontinue in vivo animal testing when in vitro testing is more than adequate for this type of use.
In addition, I know that the Center for Biologics Evaluation and Research at the FDA is planning on publishing draft guidance shortly on the use of human and animal origin components in cell and gene therapy manufacturing. I expect that they will touch further on ethical concerns linked to the use of human-derived components.
LB: In general, removing any animal- and human-derived raw materials is key. There are many recombinant sources of both sera and proteins that are available and being used in several approved drug products on the market.
In addition, thinking about sustainability, there is a trend towards using single-use materials and having single-use facilities. While these are great for reducing microbial contamination and cross-contamination, they come with the caveat of increased consumption of single-use plastics. This is a tricky situation because, while an increase in the number of single-use plastics being generated and used is not good for the environment, there is a lot of evidence to suggest that moving from stainless steel to single-use bioreactors, for example, can actually decrease carbon footprint by reducing the amount of energy, water, and cleaning solvents that are used. In addition, single-use systems, manufacturers, and users are also exploring ways in which to recycle these plastics in a safe and compliant manner.
From an end user point of view, you should do your research and look to work with suppliers that have a focus on sustainability.
KH: As the cell therapy field continues to evolve, what future trends or innovations do you foresee in raw material selection and supply chain management? How should organizations prepare for these changes?
LB: In addition to the Standards Coordinating Body’s ongoing efforts to create a standard CoA for ancillary materials, we are also going to be asking suppliers to have an identity test for all materials. This change will definitely affect suppliers—especially if they supply a complex cell culture medium, for example.
For users of raw materials, I hope that additional clarifications will be made by health authorities. Even with all of the great guidance out there, there are still some grey areas.
Lastly, these guidelines are very focused on the quality of material. They answer questions about what kind of release testing should be done, specifications, and things like that. However, there is not a lot of guidance on the actual manufacturing processes. For example, they do not address if a raw material manufacturing process needs to be validated or not. That is a small gap in information that I hope will be addressed in the future.
KK: Building on what Lili just said about standardization, I also think that we will see more standardization of critical raw material attributes and hopefully, better analytical methods to characterize complex raw materials like cell culture media or novel lipids. I think that we will also see a shift towards more defined media formulations and a move away from those high-risk components like human- and animal-derived materials in favor of recombinant or synthetic forms.
KH: In summary, it is crucial to conduct a raw material risk assessment and prioritize the critical aspects for your business. Additionally, it is important to ensure that your chosen vendors can meet these prioritized needs. Early communication with suppliers is essential to align the required documentation for regulatory filings and confirm their ability to scale up manufacturing to meet late-phase and commercial demands. Lastly, available and upcoming guidelines and risk assessment tools can be utilized to inform raw material selection and qualification decisions.
Thermo Fisher Scientific can help support raw material selection for cell and gene therapy clinical and commercial manufacturing with the Gibco CTS portfolio of products (Figure 1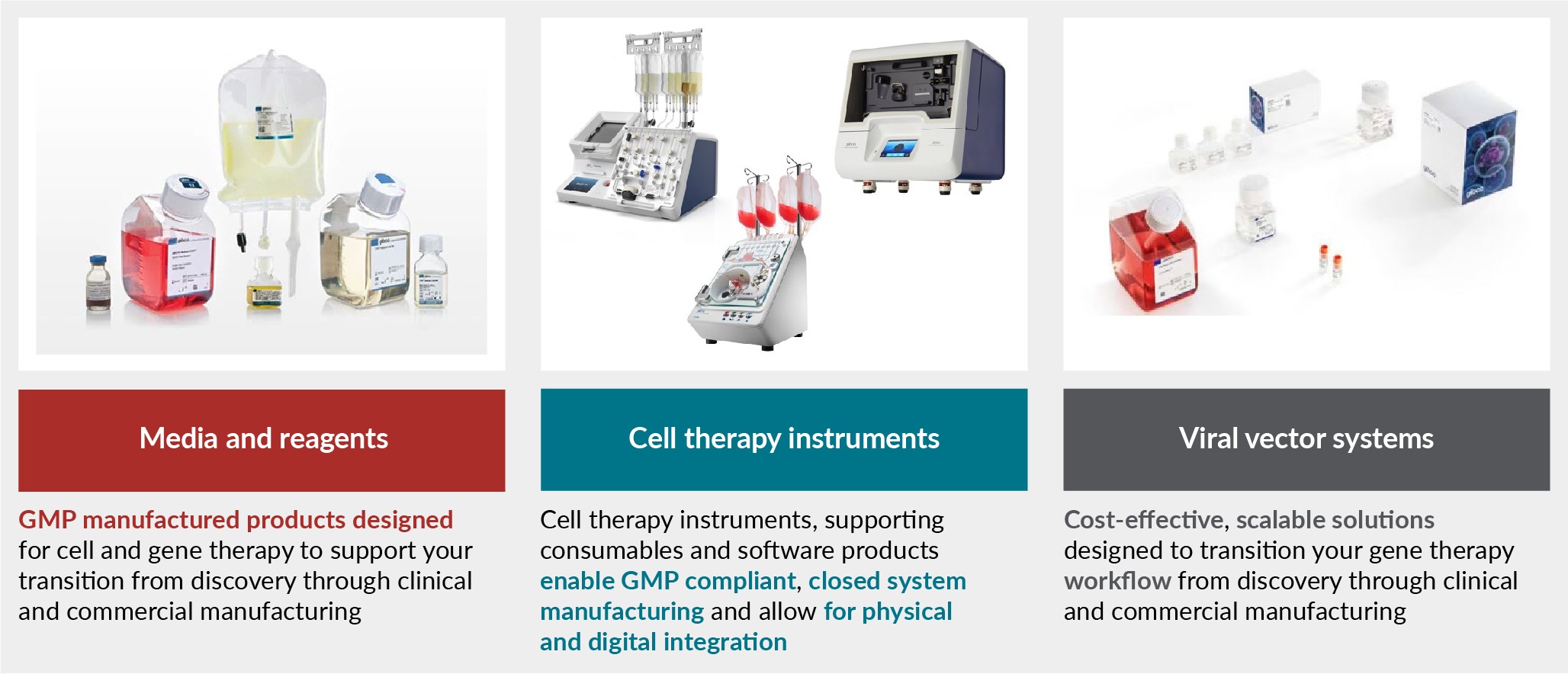 The Gibco CTS portfolio of products.).
The Gibco CTS portfolio of products.).
Gibco CTS media and reagents are GMP manufactured in ISO 13485 facilities and undergo extensive QC testing. They comply with US and EU guidelines for ancillary materials to support the transition from discovery through clinical and commercial manufacturing. Comprehensive documentation packages are available including certificate of analysis, certificate of origin, drug master file, and/or regulatory support files.
Gibco CTS cell therapy instruments include the Rotea™ Counterflow Centrifugation System, the Xenon™ Electroporation System, and the DynaCellect™ Magnetic Separation System. These instruments, as well as their supporting consumables and software products, enable GMP-compliant, closed system manufacturing and allow for physical and digital integration. As with the CTS media and reagents, the instrument consumables undergo extensive QC testing, comply with regulatory guidelines, and include comprehensive documentation packages. Additionally, a global team of Thermo Fisher Scientific cell and gene therapy field application scientists and service engineers provide comprehensive support to maximize uptime.
Viral vector systems such as the AAV-MAX Helper-Free AAV Production System and the LV-MAX Lentiviral Production System offer cost-effective, scalable solutions to support developers’ gene therapy workflows. They were designed to provide high-titers with serum-free reagents and protocols for a scalable, suspension-based platform. The viral vector multi-component systems undergo extensive safety testing, comply with regulatory standards, and include comprehensive documentation packages such as the Cell Line Documentation Package.
To learn more about our comprehensive CTS portfolio and support offering to advance your therapies, please visit www.thermofisher.com/CTS.
Biographies
Kasey Kime is a seasoned professional in global quality and regulatory affairs within the life sciences sector, with over 15 years of experience. She presently holds the role of Director of Regulatory Affairs at Thermo Fisher Scientific, where her focus is centered on technology and tools essential for cell and gene therapy manufacturing. Kasey’s specific interests lie in areas such as CMC, advanced manufacturing, and companion diagnostics. Her education comprises a BSc in Medical Laboratory Science, complemented by post-graduate degrees in Medical Microbiology, Quality Systems Management, and Business Administration. Beyond her role at Thermo Fisher Scientific, Kasey remains actively engaged by contributing to the industry, notably through her roles in the Australia and New Zealand ISCT Regulatory Committee and the Alliance for Regenerative Medicine CMC Advisory Group.
Lili Belcastro is a Senior Principal Scientist at Bristol-Myers Squibb. She leads the Material Sciences group in cell therapy development. Lili has over 15 years of experience in preclinical and clinical cancer biology, cell and gene therapy product development, and method development, working with a variety of complex biological molecules, small molecule inhibitors, ancillary materials, starting materials, and gene editing materials. Lili holds a PhD in Cancer Biology from a joint program with the University of the Sciences and The Wistar Institute in Philadelphia.
Kyle Hondorp is a Senior Manager, Product Management for the Cell and Gene Therapy at Thermo Fisher Scientific, where she is responsible for managing the Gibco™ Cell Therapy Systems (CTS™) immunotherapy and adult stem cell culture media and reagents. Kyle initially joined Thermo Fisher Scientific in 2018 as Product Manager for the Applied Biosystems™ PCR plastics and thermal cycler portfolio, after spending 18 years working for Active Motif, a life sciences company focused on providing solutions for epigenetic research. Kyle earned her BSc in Genetics at the University of California, Davis and MBA at California State University, San Marcos.
Affiliations
Kasey Kime
Director, Regulatory Affairs,
Cell, Gene, and Advanced Therapies,
Thermo Fisher Scientific
Lili Belcastro PhD
Associate Director,
Bristol-Myers Squibb
Kyle Hondorp
Senior Manager, Product Management,
Cell and Gene Therapy,
Thermo Fisher Scientific
![]()
Authorship & Conflict of Interest
Contributions: The named author takes responsibility for the integrity of the work as a whole, and has given their approval for this version to be published.
Acknowledgements: None.
Disclosure and potential conflicts of interest: The author has no conflicts of interest.
Funding declaration: The author received no financial support for the research, authorship and/or publication of this article.
Article & Copyright Information
Copyright: Published by Cell & Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0 which allows anyone to copy, distribute, and transmit the article provided it is properly attributed in the manner specified below. No commercial use without permission.
Attribution: © 2024 Thermo Fisher Scientific Inc. All rights reserved. All trademarks are the property of Thermo Fisher Scientific and its subsidiaries unless otherwise specified. Published by Cell & Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0.
Article source: Invited. This article is based on a transcript of a webinar, which can be found here.
Webinar recorded: Nov 9, 2023; Revised manuscript received: Dec 19, 2023; Publication date: Jan 5, 2024.
