
The Regulatory Environment for Cell Therapies in Australia – an Opportunity to Expedite Clinical Development
Cell Gene Therapy Insights 2018; 4(6), 523-533.
10.18609/cgti.2018.053
Early phase clinical development of cell based therapies in Australia can benefit from an abbreviated regulatory approval pathway, requiring limited resource burden relative to a European clinical trial application (CTA) or US Investigational New Drug application (IND), resulting in more rapid clinical trial initiation. With data that are generated in accordance with International Council for Harmonisation of Technical Requirements for Human Use (ICH) Good Clinical Practice (GCP), technical expectations in keeping with ICH/EU/US requirements, adherence to Pharmaceutical Inspection Co-operation Scheme (PIC/S) guidance (while requiring Good Manufacturing Practice (GMP)-like investigational product), and a government funded research and development (R&D) cashback scheme that can see tax offsets of up to 43.5%, Australia represents a highly cost-effective and efficient location for generation of clinical data that are valid for use in regulatory submissions in key jurisdictions.
Submitted for Review: Apr 13 2018 Published: Jul 17 2018
Clinical research in Australia benefits from a rapid trial approval pathway, world-class GCP trial units and clinical research organizations (CROs), experienced cell therapy manufacturing units for early stage trials, and a generous R&D tax cashback scheme intended to promote the sector. Australia is an observer of the ICH, and has regulatory standards comparable to (and accepted by) the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA). Here, we outline key considerations for Australian early access initiatives, clinical trial approval and opportunities to be leveraged to rapidly initiate clinical studies in a highly cost-effective manner.
Clinical trials & special access to cell therapies preregistration in Australia
Clinical trials of unapproved therapeutic goods in Australia are conducted under either the clinical trial notification (CTN) or clinical trial exemption (CTX) scheme, depending on the perceived risk profile and novelty of the therapy. The vast majority of clinical trials in Australia are performed using the CTN scheme, which requires a scientific and ethical review by the applicable human research ethics committee(s) (HREC(s)) only, with notification to, but not review by, the regulator, the Therapeutic Goods Administration (TGA). The HREC review typically takes 8 to 12 weeks, with a CTN notification period to the TGA of approximately 10 days. For such an application, only central trial documents are needed i.e., the clinical trial protocol, Investigator’s Brochure and patient information and consent form, with an option of an independent toxicology report to support nonclinical safety data. Submission of these documents can be to individual HRECs, or, where required, through single submission to multiple HRECs via a Human Research Ethics Application (HREA). This capitalises on the National Mutual Acceptance (NMA) of scientific and ethical review of multi-centre clinical research in all Australian jurisdictions, except Tasmania and the Northern Territory. Currently, fees are typically around AU$6000 for HREC review, plus AU$350 for TGA notification.
This abbreviated application requirement, absence of a formal regulatory review, and short assessment timeline make the CTN an attractive option for Sponsors seeking rapid clinical trial initiation, with little resource requirement for document preparation relative to other jurisdictions. Indeed, the success of the CTN is demonstrated by increasing traction among Asian countries for a similar, aligned system. Already adopted in Japan, the CTN system is under consideration in South Korea, where it is intended to recognize authorization from other regulatory agencies, and Singapore is transitioning to a risk-based CTN scheme for some trials as of 2018.
In contrast to the CTN, the CTX Scheme requires submission of a dossier in Common Technical Document (CTD) format for review by the TGA, similar to a US IND or European CTA. In this case, the evaluation process typically takes 30 to 50 working days plus clock-stops, with a fee of AU$21,100. Although accepted by all Australian clinical trial sites, an HREC approval must also accompany CTX approval from the TGA.
The choice of whether to follow the CTN or the CTX scheme lies initially with the Sponsor and then with the HREC, and depends on the biological classification (see below) and the scientific expertise required for assessment (Figure 1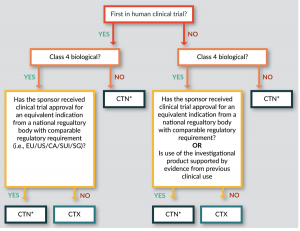
- Access to unapproved therapeutic products may be required outside of a formal research setting. In recognition of this, the TGA operates a number of additional schemes to control such access:
- Authorised Prescriber Scheme (APS) [1] authorises a medical practitioner to prescribe a specified unapproved therapeutic product to specific patients with a particular medical condition, under the conditions that the prescriber ‘is suitably qualified and trained for the condition and product, is able to best determine the needs of the patient, and can monitor the outcome of therapy.’ The prescriber must report to the TGA the number of patients treated on a 6 monthly basis. Clinical justification to become an Authorised Prescriber requires review by the applicable HREC only.
- By contrast, the Special Access Scheme (SAS) [2] refers to arrangements, which provide for the import and/or supply of an unapproved therapeutic good for a single patient, on a case-by-case basis.
Regulatory definition of cell based medicinal products in Australia
Regulation of human cell and tissue-based, or live animal organ, cell and tissue-based products in Australia is legislated in the Biologicals Regulatory Framework (BRF) [3]; its implementation is overseen by the TGA. Under the framework, a ‘biological’ is defined as:
‘an item made from or containing human cells or human tissues, or live animal organs, cells or tissues and that is used to (i) treat or prevent disease or injury, (ii) diagnose a condition of a person, (iii) alter the physiological processes of a person, (iv) test the susceptibility of a person to disease or (v) replace or modify a person’s body part(s)
Under the Therapeutic Goods Regulations 1990 (amended 2018), a ‘biological medicine’ may also be produced through recombinant technology or biotechnology. Biologicals are defined in four classes (Table 1).
| Table 1 Classification of biologicals (Australian Biologicals Regulatory Framework). | |||
|---|---|---|---|
| Class | Definition | Perceived Safety Risk | Exampls |
| Class 1 | Minimal manipulation, intended for homologous use | Very Low | No examples identified to date |
| Class 2 | Minimal manipulation, intended for homologous use | Low | Dental pulp-derived stem cells for tooth regeneration |
| Class 3 | More than minimal manipulation and/ or not for homologous use; cells processed using a method (i.e. enzymatic digestion) that has the potential to alter cells/tissue, but processing does not change the biological properties of the product | Medium | Mesenchymal stem cells for treatment of graft-versus-host disease. Mesenchymal stem cells for the repair of myocardial ischaemia. |
| Class 4 | Substantial modification of cells that alters their inherent biological properties; contains live cells, tissues or organs, HCTs with an introduced function not intrinsic to the donor, and pluripotent stem cells or products following differentiation thereof | High | Viable HCTs considered to pose a high risk due to the level of manipulation and/or current lack of safety data. Stem cells for cardiac muscular repair: stem cells isolated from bone marrow. Genetically modified cells |
Importantly, a CTX is required for all first in human (FIH) trials with a Class 4 biological product (unless a trial has been authorized for an equivalent indication in the EU, US, Canada, Singapore or Switzerland). Where Class 4 biologicals have existing clinical trial approval in other jurisdictions, or existing clinical data, and for all other classes of biologicals, a more rapid CTN approval via an HREC is appropriate.
At present, most autologous human cell therapies (HCTs), unless blood or blood components or substantially manipulated, can be supplied without approval by the TGA [4]. To meet this exemption, the donor starting material must be collected from a patient under the clinical care and treatment of a medical practitioner, and the same medical practitioner must manufacture (or oversee manufacture) and perform/supervise the use of the autologous HCT for treatment of a single indication in a single course of treatment for the same patient.
From July 2018, the BRF is expected to be revised such that regulation of autologous HCTs will be based on risk. The following changes are expected for implementation:
- Autologous HCTs will remain exempt from the BRF only where used in an accredited hospital and with no manipulation (e.g. skin grafts and haematopoietic cells for reconstitution of blood).
- Autologous HCTs with minimal manipulation will require regulation by the TGA but not under the BRF, meaning they are exempt from CTN, CTX or SAS requirements.
- Cells manufactured and used outside of accredited hospitals, cells subject to more than minimal manipulation, and cells for non-homologous use will fall under the BRF. For these products, including adipose-derived cell extract and conditioned serum, CTN, CTX, SAS or APS criteria will apply.
Therapeutic Goods Order 88 [5] describes the required standards for donor selection and testing for the generation of HCT products in Australia. These include the requirement for the person collecting or manufacturing to have procedures in place, approved by the TGA (including restrictions on donors), that demonstrate mitigation of transmission risk of infectious diseases, processes for notifying persons/organisations of evidence of infectious disease, and microbial specifications for HCT quality.
Requirements for investigational products of gene technology
A company may also need to consider the Australian Gene Technology Regulations 2001 (‘the Regulations’) [6]. Under the Australian Gene Technology Act 2000 (‘the Act’) [7], gene technology is defined as a technique that modifies genes or other genetic material, excluding sexual reproduction, homologous recombination (not involving gene technology) or other techniques as specified by the Office of the Gene Technology Regulator (OGTR). Australia currently has a ‘process’ driven trigger that defines whether a cell therapy is considered a genetically modified organism (GMO) i.e., if the manufacturing process involves gene technology techniques, it may be defined as a GMO under the Act. In contrast, countries such as Canada have a ‘product’ driven trigger, where it is the final product that is assessed as the GMO, regardless of the gene technology techniques utilized during manufacture. However, it should be noted that in Australia genetically modified somatic cells are generally exempt from regulation under the Regulations (2001; Schedule 2, Part 1(3)). Two conditions need to be met for a product to qualify as an ‘Exempt Dealing’:
- Firstly, the somatic cell therapy must not be capable of giving rise to infectious agents as a result of the modification; and
- Secondly, measures are to be taken to ensure that any recipient is not carrying a virus that is capable of recombining with the modified nucleic acid in the somatic cell therapy.
The onus is on the Sponsor to ensure that the two conditions to qualify as an Exempt Dealing are met, as the OGTR has no formal Exempt Dealing application or evaluation process. An Institutional Biosafety Committee (IBC), either independent or associated with the clinical trial site, should be notified that the product is considered an Exempt Dealing. The Sponsor should request that the IBC provide their own interpretation with justifications in a letter to be included in the HREC application to facilitate the HREC’s understanding during review of the clinical trial documentation.
Should a cell therapy product meet the definition of a GMO under the Act and associated Regulations (2001), a clinical trial application may also require a license from the OGTR. Licensable products, or ‘dealings’, are defined as either Dealings Not involving Intentional Release (DNIR; low-risk dealings that are unlikely to shed from trial participants and are not infectious) or higher risk Dealings involving Intentional Release (DIR). DNIRs take 90 working days for evaluation; DIRs take 150 (or 170) and 255 working days to evaluate for limited and controlled release and uncontrolled release, respectively. Both application types require prior vetting and review by an IBC.
Technical expectations
In keeping with its recognition of clinical trial authorisations from certain overseas regulators, the TGA seeks to closely align expectations for development of novel therapeutic products with those of comparable international counterparts. As a result, technical data requirements for Australian prescription medicine registration applications (analogous to a Marketing Authorisation Application/Biological Licence Application and New Drug Application) or post authorisation variations overlap with or incorporate the technical aspects of relevant European Union (EU) and ICH guidelines. To ensure relevance and applicability to the Australian market, adoption of EU or ICH guidelines follows extensive internal and external consultation. All EU and ICH guidelines adopted in Australia are listed online [8]. Adopted guidelines are not mandated by Australian legislative requirements, with primary governance defined by the Therapeutic Goods Act 1989 and the Therapeutic Goods Regulations 1990, and supported by additional Therapeutic Goods Orders, Notices and Determinations. Nonetheless, guidance to Sponsors contained within adopted guidelines should be followed, and any deviation from an applicable guideline relevant to an application must be justified.
Importation & GMP requirements
Importation of cell therapies into Australia requires an import permit issued by the Australian Quarantine and Inspection Service (AQIS). An AQIS permit application is completed through the Biosecurity Import Conditions System (BICON) web-based portal and takes 20 working days to process. Australia follows Pharmaceutical Inspection Co-operation Scheme (PIC/S) guidance. Therefore, investigational products in early clinical development should be manufactured to ‘GMP-like’ standards, defined as a manufacturing process where the quality of material produced is equivalent to that manufactured to GMP standards but where the manufacturing facility itself does not necessarily need GMP certification. Practically, GMP-like investigational products may lack the same extent of process and procedure validation required for GMP manufacture. Nonetheless, materials of sufficient quality are used and suitable testing is performed (according to ICH, EMA or FDA guidelines), to ensure the production of a safe and effective investigational product with suitable stability for the clinical trial. GMP compliance may not be subject to inspection until an application for product registration, and there seems to be limited evidence of GMP licensure being mandated by the TGA during clinical trials, but safety reporting to the TGA during the trial will be expected according to GCP. Given this GCP expectation, trial data obtained from studies in Australia, with fast-track start-up given ‘GMP-like’ manufacturing requirements, is considered valid for subsequent European and US applications.
It should be noted that whereas a full Module 3 (Quality) and Module 2.3 (Quality Overall Summary) will be required in CTD format for CTX applications, only details of the pharmaceutical and chemical properties, formulation, stability and shelf-life, stated in the Investigator’s Brochure, are required for a CTN.
Comparing clinical trial timelines between australia & the US/EU
Clinical trial timelines, including procedures and requirements, are outlined in Table 2. Australia provides a number of benefits to cell therapy manufacturers, encouraging rapid trial start up.
| Table 2 Comparison of clinical trial application requirements and timeline. | |||
|---|---|---|---|
| Class | Australia | EU | US |
| All products> | |||
| Application process |
|
|
|
| Typical timeline/ resource requirement for document preparat |
|
|
|
| IP Grade | GMP-like* | GMP | GMP |
| Timelines for approval** | Ethics:
| Ethics:
| Ethics:
Regulatory:
|
| * As stated in the BRF 2018, GMP licensing or certification of manufacturing sites is required, unless the persons or goods are exempt e.g. first in human trials. However, there seems to be limited evidence of GMP licensure being assessed or enforced by the TGA during clinical development of biological (and non-biological) investigational products. ** GMO investigational products are assessed for potential environmental risk (primarily) in the different jurisdictions by different bodies. In Australia, the evaluation is performed by the OGTR, unless the product is an Exempt Dealing. For GMOs that are not Exempt Dealings, this evaluation process takes 90 to 150 working days, depending on the perceived level of risk of the GMO, before the clinical trial can commence. ADME: Absorption, distribution, metabolism, and excretion; CMC: Chemistry and manufacturing controls; CTA: Clinical trial application; CTN: Clinical trial notification; CTX: Clinical trial exemption; GMO: Genetically modified organism; IB: Investigator’s Brochure; IMPD: Investigational Medicinal Product Dossier; IND: Investigational new drug application; IRB: Institutional review board. | |||
Research and development (R&D) tax incentive
In addition to an abbreviated regulatory pathway under the CTN which can lessen resource burden for document preparation and expedite trial initiation, the Australian government currently offers the R&D Tax Incentive to eligible parties. The incentive, which is intended to stimulate and support innovation for Australian businesses, provides a targeted tax offset to encourage companies to engage in R&D. If medicinal product developmental activities are performed within Australia by an Australian business, the cost of the activities should be eligible for a R&D tax rebate (43.5% at the time of publication). Specifically, the commercial entity claiming the rebate is required to be incorporated under a foreign law, to have at least AU$20,000 of eligible expenditure on eligible R&D activities, and to be both a resident of a country with which Australia has a double tax agreement that includes a definition of ‘permanent establishment’ and carrying on business in Australia through a permanent establishment as defined in the double tax agreement.
The scheme enables a 43.5% refundable tax offset for companies if they have an aggregate annual turnover of less than AU$20 million and a non-refundable 38.5% tax offset for all other eligible companies. For foreign entities that establish a presence in Australia and meet other eligibility criteria, the scheme affords conduct of highly cost-effective clinical trials.
Clinical experience in Australia with cell based therapies
Given the attractive benefits which include an abbreviated regulatory pathway, the ‘GMP-like’ manufacturing requirement and R&D cashback incentives, how experienced is the Australian clinical environment with cell based therapies and advanced therapy medicinal products?
For confidence in GCP trial conduct, each Australian state capital (Adelaide, Brisbane, Melbourne, Perth, Sydney) accommodates a dedicated Phase I unit. Furthermore, dedicated cell therapy facilities, such as the Victorian Comprehensive Cancer Centre (VCCC) at the Peter MacCallum Cancer Centre in Melbourne, enable on site integrated cell processing capabilities with patient access. This has facilitated, for example, the FIH study of CAR-T cells targeting the Lewis Y antigen in patients with refractory acute myeloid leukaemia (AML) and solid tumours.
Review of the Australia New Zealand Clinical Trials Registry (ANZCTR) and ClinicalTrials.gov, for clinical trials currently active in Australia with cell based medicinal products, identifies a predominance of oncology trials with somatic cell therapy medicinal products being most common (Figure 2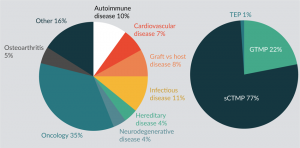
Translational Insight
Australia represents an opportunity for manufacturers of cell based medicinal products to rapidly enter the clinic and generate early phase clinical data. Key advantages/disadvantages for Sponsors considering the region are presented in Table 3.
| Table 3 Advantages and disadvantages of clinical trials for cell based medicinal products in Australia. | |
|---|---|
| Advantages | Disadvantages |
|
|
|
|
|
|
|
|
| |
| |
| |
| |
| CTA: Clinical trial application; CTN: Clinical trial notification; ICH: International Council for Harmonisation of Technical Requirements for Human Use; IND: Investigational new drug application; GCP: Good Clinical Practice; GMO: Genetically modified organism; OGTR: Office of the Gene Technology regulator. | |
Financial disclosure/acknowledgements
Authors are employees of Clinical Network Services, an Australian clinical Contract Research Organisation.
References
1. www.tga.gov.au/form/authorised-prescribers
2. www.tga.gov.au/form/special-access-scheme
3. www.tga.gov.au/regulatory-framework-biologicals
4. Therapeutic Goods Order; (TGO), No. 1 of 2011, Item 4(q) https://www.tga.gov.au/therapeutic-goods-excluded-goods-order-no-1-2011
5. www.legislation.gov.au/Details/F2013L00854
6. www.legislation.gov.au/Details/F2016C00615
7. www.legislation.gov.au/Details/C2016C00792
8. www.tga.gov.au/publication/scientific-guidelines
Affiliations
Simon Bishop Clinical Network Services (CNS) Pty Ltd Level 4, 88 Jephson Street, Toowong, QLD 4066, Australia
Simone Flight Clinical Network Services (CNS) Pty Ltd Level 4, 88 Jephson Street, Toowong, QLD 4066, Australia
Natalie Thomas Clinical Network Services (UK) Ltd, Ground Floor, 20 The Causeway, Bishop’s Stortford, Hertfordshire, CM23 2EJ, UK