
Driving CAR-T Manufacture Optimization through Technology Innovation
Cell Gene Therapy Insights 2018; 4(4), 317-325.
10.18609/cgti.2018.036

In general, which parts of the CAR-T manufacturing process can be automated or semi-automated?
Critical parts of the CAR-T cell manufacturing process can be automated now and there is an urgency for the industry to proceed. It is important to note that approximately 5 x 109 T cells are typically harvested in a single leukapheresis from a healthy donor. This number is dramatically greater than the number of genetically modified T- cells actually infused into patients. (Novartis’s Kymriah™: 0.2 to 5.0 x 106 per kg and Kite’s Yescarta™: 2 x 106 per kg). Even more disturbing, the low numbers of infused cells follow cell expansion periods of a week or more, which highlights the tremendous losses of T cells that routinely occur within the current manual manufacturing processes.
Losses of critical cells occur throughout the manufacturing process, but perhaps the most punishing losses occur in the first stage, obtaining a purified MNC fraction from the patient’s collected blood cells so that efficient isolation of T lymphocytes can take place. We have been informed by laboratory personnel routinely manufacturing CAR-T cells that current manual methods can result in losses of 50 to 90% of the target cells, so pursuing automation for this step is extremely important.
Successful automation must minimize the loss of MNCs while depleting platelets, red cells, granulocytes and blasts and washing of DMSO (from previously frozen units) to provide the purified mononuclear cell fraction for efficient antibody selection of the T-cells. Successful automation must also overcome the significant complications that result from the several ways in which these patient cells are received at the manufacturing facility: (peripheral blood or leukapheresis, fresh or frozen)
The next step of the CAR-T manufacturing process requiring automation is the selection of T lymphocytes from the MNCs. An automated selection step must obtain a pure fraction of T cells in order to maximize efficiency of downstream processes. Residual monocytes remaining in the T lymphocyte fraction after antibody selection can lower transduction efficiency and subsequent expansion of T cells. Further, if the presence of monocytes is significant, as it often is after conventional magnetic bead-based selection of T-cells, manual methods of trying to deplete those monocytes lead to unavoidable further losses of target T cells.
Once the pure T-cell fraction is obtained, successful automation of activation must overcome additional complications. For example, some magnetic bead products used for activation are themselves a problem sufficient that the FDA requires the clinician to count them in (typically more beads than the number of cells to be activated) and then, count them out after multiple washing steps. Understandably, the FDA does not want those cell-sized beads remaining in a product that will be transfused into the patient.
What advice would you give to companies considering incorporation of automation, and when ideally should they be looking to expect a return on investment?
If widgets were what was being manufactured, then a simple return on investment (ROI) analysis would suffice. But we are not making widgets, we’re trying to assemble a sufficient number of viable, genetically modified T-cells to save a life, so patient survival dominates the calculation. However, it is useful to acknowledge the manufacturing model provided by Silicon Valley: as chip manufacturers upgrade the automation of new chip designs to obtain greater numbers, costs drop and quality improves.
Even though sick patients provide starting cell populations that are greatly variable in number and quality, a complication not faced by chip manufacturers, automated devices in this field will still provide the desired cost reductions as long as every automation step adopted not only reduces time to manufacture but also achieves improvements in cell recovery or viability or purity and/or consistency.
At this time, we are unable to simply place a collection of cells from a patient in one end of an automated system and, shortly thereafter, have a transfusion ready CAR-T cell product come out the other end. But, systematically introducing modular automation into the manufacturing process as soon as practical will streamline the process now and make the final transition to a fully automated process more realizable later.
What kind of performance can be expected from the CAR-T express system compared to non-automated manufacturing methods?
We believe our automated modules, when compared to legacy manual systems, will provide performance gains in purifying MNCs, washing contaminants from cell fractions, antibody selection and activation of T-cells while significantly shortening processing times.
The literature is clear that isolation of clinically relevant numbers of MNCs from peripheral blood, with Ficoll, results in losses >30% of target MNCs and requires ~4 hours. Our X-LAB™ automated, closed system provides consistent MNC recoveries >90% and depletion of red blood cells, granulocytes and platelets in approximately 30 minutes.
Again, the literature is clear that conventional manual, or 1st Gen. mechanical, methods of washing thawed leukopaks, results in losses of >30% of the T-cells. In contrast, the X-WASH™ automated system routinely provides >99 % depletion of DMSO with losses of 85%, which is typically 10–15% greater than the levels obtained through traditional magnetic beads.
At the downstream expansion step, our experiments to date document that bubble-activated cells are consistently activated and expand as well or, more often, better than T cells isolated from the same starting material by conventional magnetic bead technologies.
At a minimum, we expect our modular, automated technologies will provide at least a 10–20% more efficient process at every manufacturing step than the existing manual processes used right now.
At this stage, our data is largely based on healthy donor cells but we anticipate being able to derive new comparative data from ‘real world’ patient samples in the near future. It is of course challenging to obtain significant numbers of cells from sick patients with which to test a new technology. The priority for these often incredibly sick patients is to obtain as many viable cells as possible for their CAR-T treatment and not for research purposes. However, we are working with organisations that, periodically, have access to ‘excess’ frozen patient cells – circumstances in which sadly the patient may have passed away before the cells were processed, or on the rare occasion that the patient provides an excess of cells. Given these cells are frozen and from sick patients, we will be able to test our technology on some of the ‘worst’, but realistic, conditions over the next few months and we are hopeful the performance tracks the performance with cells from healthy donors.
Is the platform scalable and does it require extensive customization?
The CAR-TXpress platform products are scalable, and do not require extensive customisation. In fact we’ve designed each module (X-LAB, X-WASH or X-BACS) to require only programming of the control modules. For example, if purified MNCs are desired, the control module program will vary depending on whether your starting material is leukapheresis or peripheral blood. But once the control modules are programmed, the CAR-TXpress systems should repeat the process consistently.
As a test of scalability, we set up a competition at a local GMP cell processing laboratory to obtain activated T cells from a unit of peripheral blood. The blood was divided into two equal samples and one competent laboratory person, performing conventional manual processes with Ficoll and magnetic beads to isolate and activate T-cells began working while an equally competent laboratory person, starting at the same time, utilized the X-LAB and X-BACS to accomplish the same goals. The 2nd laboratory person, using the CAR-TXpress sytems, had purified the MNCs and selected and activated the CD3+ cells before the 1st lab person, using conventional means, had even completed the Ficoll purification of their MNCs.
We’ve designed the CAR-TXpress system to be modular, which in itself helps companies with cost efficient scaling, because a single, expensive “swiss army knife” type of device is not tied up weeks. With a modular platform, you can design your workflow to be as efficient as possible.
Further, if significantly fewer T lymphocytes are lost during the MNC purification, selection and activation steps, then it might be possible, at the completion of transduction, to simply wash those genetically modified cells and transfuse them into the patient, letting expansion occur in the patient. That strategy, if proven clinically, would drastically reduce the elapsed time from receiving the source cells from the patient to clinically administering the gene modified CAR-T cells back to the patient.
The CAR-TXpress system does not use Ficoll or magnetic beads for isolation and selection. What was the rationale for removing these methods?
Ficoll density separation of cells is a forty years-old, time consuming, manual and inefficient process to purify MNCs from peripheral blood or leukapheresis. Performing it is a rite of passage for all laboratory technicians. Your readers will all be aware of its’ many shortcomings. Most consequential, for patients requiring CAR-T cell therapy, are the typical losses of 30 to 60% of the target MNCs in this first step of the manufacturing process. These lost MNCs, ~50% of which are T lymphocytes, are never recovered and patients requiring CAR-T cell therapy can ill afford their loss.
The magnetic beads used for antibody separation of T lymphocytes from the MNCs have been used for around 25 years and have been an invaluable tool; however, challenges arise when you consider relying on them for scaling-out the manufacture of cell therapies to meet the demand of the large patient populations in need as they exhibit low throughput and poor cell recovery and purity. And, as previously mentioned, some magnetic bead products used for activation are themselves a problem sufficient that the FDA requires the clinician to count them in (typically more beads than the number of cells to be activated) and then, count them out after multiple washing steps.
In contrast, our lipid shell microbubbles simply outgas after cell selection and activation occurs. Any benign residue left is automatically removed during subsequent washing steps required after transduction and/or expansion.
Can you describe how BACS works, and how it compares to other cell separation methodologies?
Our buoyancy activated technology utilizes lipid shell, streptavidin coated bubbles, 2–3 micrometers in diameter in combination with biotinylated antibodies to select, or activate, or simultaneously select and activate, target cells, such as T lymphocytes.
In general, microbubbles are very well understood by the FDA, because they are FDA approved and routinely used for diagnostic imaging. They are directly transfused into the vasculature of the patient, distribute throughout the body and, at any given site, the right ultrasonic signal will cause them to implode inside the body, thereby broadcasting a signal that provides an image of the nearby tissue, so that the clinician can non-invasively “see” this tissue.
Conventional cell selection technology utilizes magnetic beads coated with antibodies to attach to surface antigens on the target cells and then passages the cells past a magnetic field to trap the target cells and allow the non-target cells to pass. With this technology, both the target and non-target cells flow downward through the very narrow space required to achieve a uniform magnetic field. The non-target cells, unattached to magnetic beads, are expected to continue down through this tight space and into a container to be removed; however, the intermingling of target and non-target cells concentrated together as they passage this narrow space is one of the limitations of this process as non-target cells can be trapped by the target cells, thereby reducing the purity of the final product, often requiring an additional step to remove the contaminating monocytes before transduction and expansion of the T cells.
In contrast, CAR-TXpress technology bubble/antibody complexes bind to the target cells and alter their collective density so they now gently rise under centrifugation in the opposite direction of the descending non-target cells (Figure 1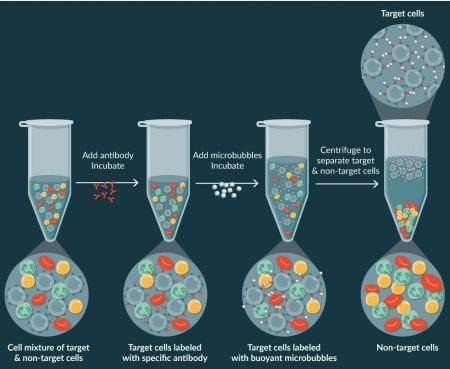
Further, our platform automates this process by utilizing a special cylindrical cartridge with a conical processing chamber in which, during centrifugation, the target cells attached to the microbubbles gently rise to the top surface of the plasma, recovering > 85% of the T cells while depleting >99% of the monocytes.
This is important because every T-cell retained in these initial processing steps will not have to be replicated through expensive and time-consuming cell expansion. Equally important, if activation of the T cells is also desired, this can be accomplished during the selection process with the X-BACS technology, by linking both CD3+ and CD28+ antibodies to the microbubbles (Figure 2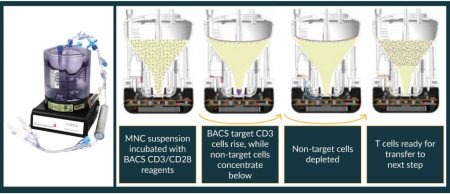
We believe the X-BACS cell separation and activation technology has distinct advantages over conventional magnetic bead technology, one of which is that lipid shell microbubbles simply outgas in a few hours and do not need to be counted in or counted out.
We believe CAR-TXpress is a breakthrough technology that has the potential to streamline the manufacturing process and we’re looking forward to moving this forward with our partners.
Are there any regulatory hurdles that may be encountered should a therapeutic company looks to incorporate the BACs process and technology into their manufacturing workflow?
In every country, of course, there are regulatory hurdles to overcome but they are not insurmountable and the best approach is to establish a Master File. For example, ThermoGenesis will maintain a Device Master File that is focussed on the X-LAB, X-WASH and X-BACS processing cartridges, within which MNC purification and target cell selection and activation occurs. This Master File will contain all the relevant information that the FDA will need to allow a Principal Investigator to include these systems in an IND.
The X-BACS reagent kits will also be supported by a Master File with all the relevant information on the antibodies, buffers and microbubbles that the FDA will need to allow a Principal Investigator to include the reagent kit in an IND.
These Masterfiles will also provide information on any residual materials from the X-BACS reagents that remain in the product at the time these cells are introduced to the patient. The microbubbles are manufactured solely from benign material that nevertheless should be reduced in the course of subsequent processing (transduction, washing, expansion and more washing). We expect these Master Files will be available to principle investigators by early 2019.
There’s a lot of people talking about high risk if you only have single source of your materials. What sort of steps do you think a manufacturer should take to try and mitigate risks around these?
First it should be understood that our products will be an addition to all the existing means of performing these cell processing steps, surely a comforting thought to potential users. Further, ThermoGenesis has more than 30 years-experience manufacturing medical cell processing devices which are re-usable multiple thousands of times. So, there will be no need for, so no burden to manufacture, large numbers of these electro-mechanical devices.
More important, the supply of single-use cartridges and single-use reagent kits used with our electro-mechanical devices will be reliably available as we are establishing long-term supply agreements with very large and reputable vendors who make the cartridges and reagents under our direction and have disaster and risk mitigation plans within the organisation.
Affiliation
Philip Coehlo
Founder and Chief Technology Officer of ThermoGenesis Corp., the device Division of Cesca Therapeutics.
![]()

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.