
Overcoming Challenges for Engineered Autologous T Cell Therapies
Cell Gene Therapy Insights 2018; 4(4), 173-186.
10.18609/cgti.2018.014
Axicabtagene ciloleucel is an engineered autologous anti-CD19 chimeric antigen receptor T cell therapy being developed for patients with refractory aggressive B-cell lymphoma. The product is manufactured in a central facility from cellular starting material containing a patient’s own T cells into which a chimeric antigen receptor transgene is directly introduced. This cellular starting material is highly variable from donor to donor and provides the single largest source of variability associated with the production process. Nonetheless, a robust manufacturing and distribution process was developed based on process understanding and appropriate process controls. Process characterization revealed process parameters that affect quality attributes, allowing appropriate control measures to be implemented. Process comparability criteria were also established as another mechanism to ensure process consistency as new manufacturing sites were introduced. A relationship between cellular characteristics of the incoming cellular starting material, cell growth and performance during manufacturing, and the ultimate product characteristics after administration to patients is also beginning to come into focus.
The adoptive transfer of T cells genetically engineered to express a chimeric antigen receptor (CAR) has achieved significant progress in treating malignant diseases. CARs are synthetic immunoreceptors whose extracellular domain is typically an antibody-derived single-chain variable fragment (scFv) that recognizes a tumor cell surface protein. The scFv is linked to intracellular signaling components that play a critical role in T cell activation, proliferation, persistence, and cytotoxicity. The first CAR T cell trials in cancer patients addressed advanced epithelial ovarian carcinoma and metastatic renal cell carcinoma and targeted the folate receptor and carbonic anhydrase IX (CAIX), respectively [1,2]. These studies were followed by studies in patients with neuroblastoma and follicular lymphoma [3,4]. Recent clinical success, however, has been achieved with CD19‐specific CAR T cells targeting B‐cell malignancies [5–10].
Axicabtagene ciloleucel (axi-cel) is an autologous anti-CD19 CAR T cell product, originating from work conducted at the Surgery Branch of the National Cancer Institute (NCI; Bethesda, MD). The CD19-specific CAR of axi-cel comprises an extracellular scFv specific for CD19 and the signaling domains of CD3ζ and CD28 and was first described by Kochenderfer et al. in 2009 [11]. The initial studies demonstrated that primary human T cells expressing this CAR could produce cytokines specifically in response to CD19+ target cells and efficiently kill primary chronic lymphocytic leukemia cells in vitro. Subsequent studies showed the potent antilymphoma activity of anti-CD19 CAR T cells together with the expected on-target/off-tumor effect of normal B-cell aplasia. These preclinical studies laid the groundwork for the first clinical report to describe successful anti-CD19 CAR T cell therapy [12].
The cell production method used to support initial trials at the NCI relied on numerous manual, open-process steps, human serum to support T cell growth, and extended cell culture to achieve a clinical dose. In those studies, freshly prepared cells were administered to patients at the same institution where the cellular starting material was collected and final product cells were prepared. This approach limited the ability to support large multicenter clinical trials, as well as scale-out for commercial cell production. Success closing some process steps such as T cell transduction with viral vectors in bags had been reported [13,14], but it was unclear if the process could be shortened and whether human serum could be eliminated from the T cell culture medium. Therefore studies were completed to simplify, streamline, and optimize the production process by removing human serum from the process to minimize the risk of viral contamination, moving process steps from an open system to functionally closed-system operations to minimize the risk of microbial contamination and standardizing additional process steps to improve process consistency [15,16].Those studies led to establishment of a simple, robust process that was suitable to support multicenter clinical trials and meet the demands for commercial manufacturing with an overall turnaround time of approximately 17 days. A schematic overview of the approach for production and distribution of axi-cel is highlighted in Figure 1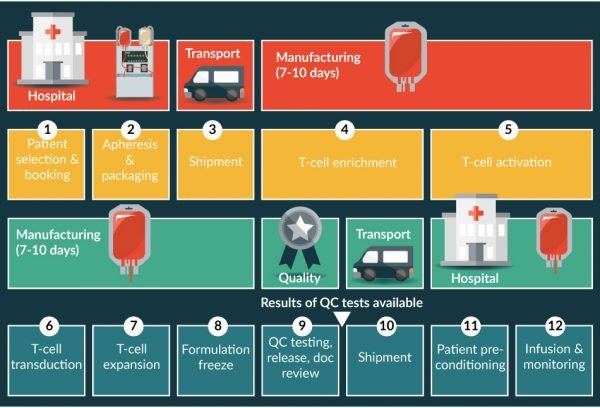
In the present study, robust manufacturing is demonstrated based on process understanding and appropriate process controls. Process characterization revealed process parameters that affect quality attributes, allowing introduction of appropriate control measures. Process comparability criteria were established based on process knowledge to ensure process consistency as changes were introduced that could unexpectedly alter product-quality attributes if not properly controlled. In addition, relationships between cellular characteristics of the starting material, cell growth and performance during manufacturing, and the final product characteristics after administration have been explored.
Production of axi-cel
For patients with diffuse large B-cell lymphoma, treatment with a single infusion of axi-cel is capable of inducing a complete remission in many cases. The response observed in one such patient; in this case, a 62-year-old who had failed prior therapy with R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone), R-GDP (rituximab plus gemcitabine, dexamethasone, and cisplatin), R-ICE (rituximab plus ifosfamide, carboplatin, and etoposide), and R-lenalidomide, but experienced a complete response after a single infusion is illustrated in Figure 2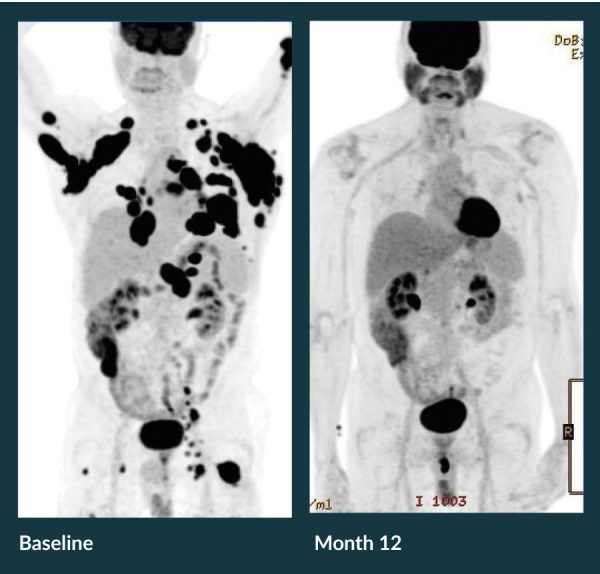
The road to commercial development of a product such as axi-cel is complex. In addition to the clinical development path, a robust and reliable commercial manufacturing process must be secured, and a commercial-ready manufacturing facility must be built and validated. Toward this latter goal, Kite commissioned a facility near the Los Angeles International Airport with a modular design to allow for scalable, efficient manufacturing that can be quickly expanded to meet demand. Close proximity to the airport helps ensure that the unique logistical requirements for product distribution can be met. The challenges associated with manufacturing and distribution of CAR T cell products were recently well described [19]. Production of axi-cel is designed to harness the power of a patients’ own T cells. The manufacturing process involves:
- Harvesting T cells from the patient’s blood
- Genetically engineering T cells to express cancer-specific receptors
- Increasing the number of engineered T cells
- Infusing the functional cancer-specific T cells back into the patient [Figure 1].
Axi-cel is manufactured from an individual patient’s blood cells obtained using a standard leukapheresis procedure. This apheresis material is placed into a validated shipping container at the collection site for transport at 1–10°C to the central cell-processing facility. After receipt, inspection, and release of the apheresis material for manufacturing, all further process steps are conducted in an International Organization for Standardization (ISO) 7 cell culture suite containing an ISO 5 biological safety cabinet and other equipment. The T cell-containing peripheral blood mononuclear cell fraction is enriched using Ficoll-based separation on the Sepax®2 instrument (Biosafe America, Houston TX) using a standard aseptic tubing kit. If required, to accommodate the cell concentration and collected volume from an individual donor, a volume reduction step is included before density-gradient separation. T cells in the peripheral blood mononuclear cell fraction are cultured in serum-free media and activated with anti-CD3 antibody and recombinant human interleukin-2.
After T cell activation, the anti-CD19 CAR gene is introduced into cells by retroviral vector transduction. Activated T cells are transferred to a cell culture bag that has previously been coated with RetroNectin® (Takara Bio USA, Inc, Mountain View, CA), and are subsequently incubated with retroviral vector. After transduction, T cells are expanded to achieve a patient dose and then washed and cryopreserved to generate the final product. A predefined CD4+/CD8+ ratio is not required for the final product [20]. After passing release tests for microbiological safety, potency, viability, purity, appearance, and identity, the cryopreserved product is shipped back to the site in a liquid-nitrogen dry shipper, and the patient receives his/her engineered T cell product after nonmyeloablative chemotherapy conditioning.
T cell activation, transduction, growth, and final formulation are all critical to an efficient manufacturing process. Because each autologous product lot is unique to a cancer patient, a robust and well-controlled process that has a very high likelihood of successful completion is essential. Axi-cel manufacturing is a continuous process with no complex downstream purification steps.
To ensure that a lot can be manufactured for essentially all patients, it is extremely important to understand the sources of process variability. The commercial axi-cel process was designed based on knowledge gained through initial process development both at the NCI and Kite [15,16], through scale-up/scale-out activities during development and through clinical experience with the ZUMA-1 trial [17,18]. In addition, process characterization was completed to understand the impact of process parameters on quality attributes (see below). After this process characterization campaign, a control strategy was developed for which appropriate operational and in-process controls were implemented. Some of the known sources of process variability included raw materials and reagents, operator activities including manual process operations, single-use components, equipment performance, and the analytical methods used to measure and test in-process and final product samples. Variation around each of these sources can be controlled to a large degree, but the largest source of variability has proven to be the donor-to-donor variability of the patient’s starting apheresis material.
Life cycle approach to axi-cel process validation
Kite has used a 3-stage life cycle approach to axi-cel process validation based on the concepts outlined in the 2011 guidance documents provided by the US Food and Drug Administration [21] and also aligned with similar European Medicines Agency guidance [22]. Process validation is defined as the collection and evaluation of data, from the process-design stage through commercial production, which establishes scientific evidence that a process is capable of consistently delivering quality product. Process validation involves a series of activities occurring during the life cycle of the product and process. Activities occurred in 3 stages: process design, process performance qualification, and continued process verification. A risk-based approach to process characterization by unit operation was used [Figure 3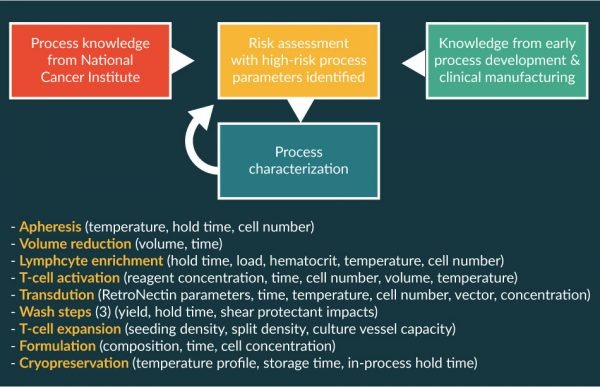
As part of process characterization, process parameters were evaluated for each unit operation as outlined in Figure 3 to identify which could be classified as critical and noncritical. Most process parameters did not affect the overall process across the tested ranges. However, a few performance parameters were very sensitive to the operating conditions, and small perturbations had substantive effect on CQAs. In those cases, the overall process-control strategy needs to ensure that CQAs stay within well-established acceptable ranges. Operating ranges for conditions such as temperature and time are straight-forward to assess across reasonable ranges. Other attributes can require more complex analysis to evaluate. An example of a noncritical process parameter is shown in Figure 5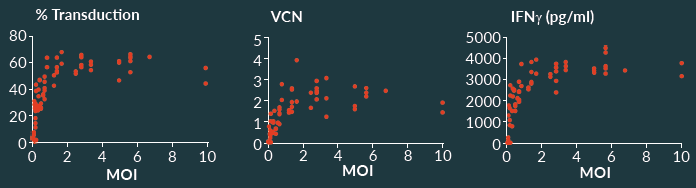
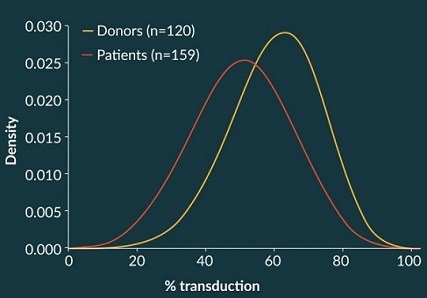
By contrast, the total viable cell concentration during certain process steps proved to have a high impact on an important CQA, namely percent transduction. This is illustrated in Figure 6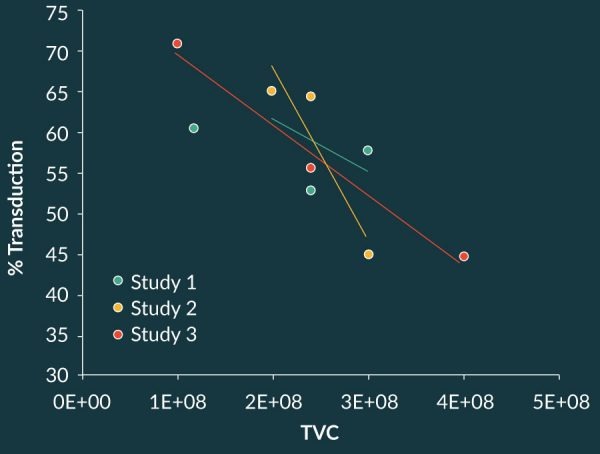
Process comparability
The manufacturing process used to support initial clinical development at the NCI [11] is now referred to as the CLP 1.0 process. Work conducted collaboratively between Kite and the NCI subsequently showed that it was possible to remove human serum from the manufacturing process stream and that many unit operations could be conducted in closed systems [15,16]. Studies using split starting material demonstrated that product manufactured using the CLP 1.0 process and the new process were highly similar. Those process improvements were then incorporated in the NCI clinical development program as process CLP 2.0, and clinical studies with patient lots prepared with the CLP 2.0 process confirmed clinical activity [23]. To support a multicenter clinical trial of axi-cel, this process was then transferred to a contract manufacturing organization (CMO) under Kite’s control. The process at the CMO was designated CLP 2.2 to indicate that processing would be conducted with apheresis starting material that was sent to the CMO for central processing and that minor process improvements to increase cellular wash recovery and ensure process integrity were incorporated. With minor differences now in place, were the 2 processes (CLP 2.0 and CLP 2.2) comparable? Studies were therefore designed to assess whether CQAs were within acceptable limits of variation when the process was conducted at the 2 manufacturing sites. Further complexity was added when Kite developed its own clinical manufacturing facility in Santa Monica, CA, and then subsequently built a commercial facility in El Segundo, CA. Was product produced at each of these sites comparable?
Two approaches have been used to assess comparability and answer this question:
- Equivalence, performance with split starting material
- Expectation, performance within prespecified tolerance intervals
In each case, comparability included demonstration that process parameters met expected established ranges. These approaches are not strictly exclusive, and a complete analysis may contain elements of both the equivalence and expectation approaches. Certain objective criteria were identified to assess comparability. To begin, a risk assessment was performed to determine which CQAs should be considered as most likely to be relevant to demonstrate product and process comparability. Percent transduction is used to calculate a patient dose, and process characterization studies proved that this attribute is highly dependent on the starting apheresis material. In this regard, percent transduction was an excellent choice for comparability assessment. Other useful CQAs included product potency to show that the transduced T cells are functional, process-related impurity clearance to assess the process capability, and general safety tests (sterility, for example) to demonstrate process integrity. Other measures of process performance were also assessed. For example, wash-step recovery, cell viability at selected process steps and at harvest, and fold-expansion of the T cell cultures each provided information to assess overall process comparability.
Using split apheresis studies, i.e., the equivalence approach, it was possible to demonstrate process comparability between the CLP 2.0 process that was being executed at the NCI and the CLP 2.2 process at the CMO. Four cellular starting materials were prepared at the NCI, and a portion of the cells were sent to the CMO. The processes were executed at each site and percent transduction, growth performance, and other process and product characteristics were assessed. Notwithstanding the inherent differences seen across the four starting materials, cell growth for each pair performed quite similarly at the respective site, as shown in Figure 7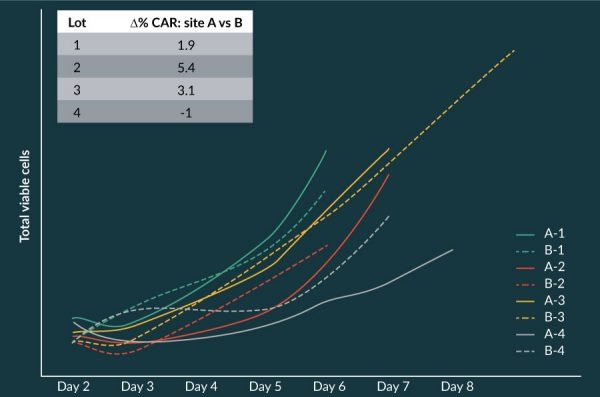
Cross-site data for percent transduction were evaluated statistically using the two one-sided test as a measure of comparability. Results demonstrated comparability at a calculated power of 89% to a p-value of 0.0142 at the very stringent percent acceptable difference of 10%.
For subsequent site transfers, either the equivalence or expectation approach has proven useful to show that the processes at each site are comparable. This risk-based assessment of comparability, based on a very clear understanding of CQAs from process characterization, provides tremendous value as products move from clinical development toward commercialization.
Clinical manufacturing correlates
The axi-cel manufacturing process has proved to be quite robust and tolerant to operating ranges across most process parameters. During execution of the ZUMA-1 clinical trial that evaluated performance in 101 clinical patients, the manufacturing process also proved to be robust across the widely heterogeneous incoming apheresis material. The percentage of T cells in the starting material varied widely [Figure 8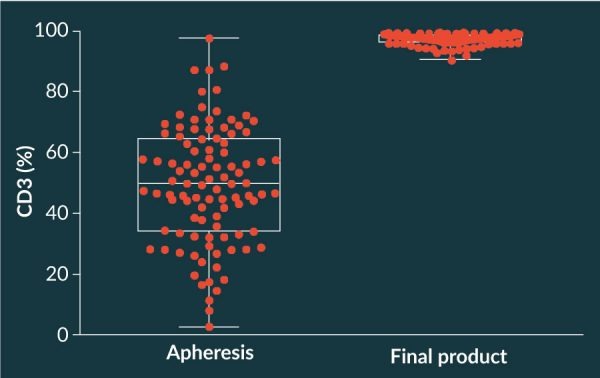
One interesting observation from the manufactured products was that the T cell-immunophenotype of the final product had an apparent more naïve phenotype than did the incoming apheresis starting material. This is illustrated in Figure 9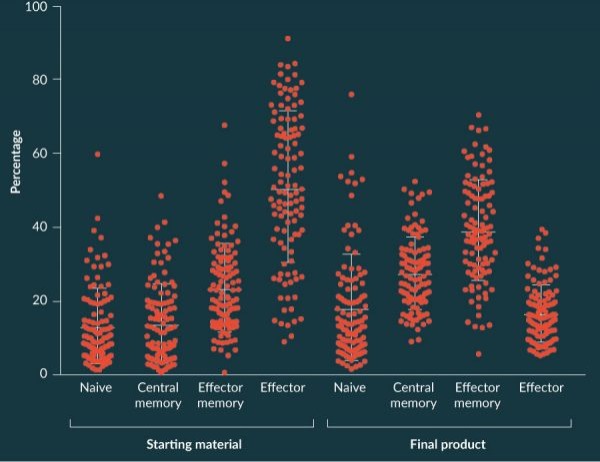
The number of infused T cells with a naïve phenotype previously have been shown to directly correlate with subsequent CAR T cell levels in the blood of patients [24]. In addition, a correlation was observed between the doubling time of T cells during production of axi-cel during manufacturing and the subsequent CAR T cell peak level in treated patients [Figure 10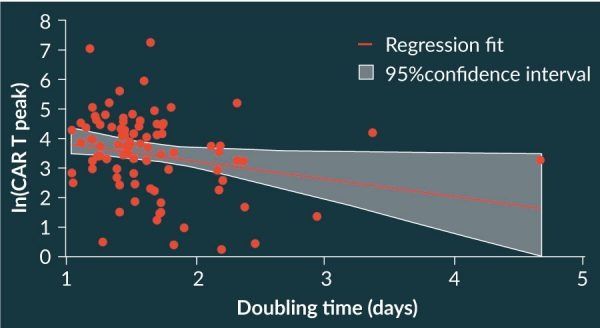
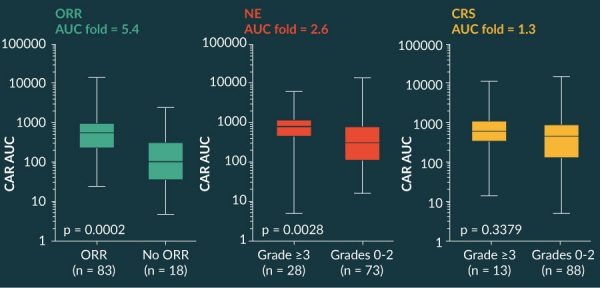
Other interesting observations relating performance during clinical manufacturing and outcomes in patients have been described [24]. Across the spectrum of clinical product lots, the CD4/CD8 ratio in the ZUMA 1 study was highly variable, with a median of 0.9 (range, 0.03–5.8). There was no difference in objective response rate, complete remission, grade >3 neurotoxicity or >3 cytokine release syndrome across the four quartiles. These data support consistent efficacy and safety across the range of CD4/CD8 in the product.
Conclusions
Production of CAR T cells for clinical and commercial applications is complex but highly feasible, and many of the general principles of drug development that have proven successful to deliver well-characterized biologics are applicable for cell therapy products. Most interestingly, robust manufacturing is feasible even with highly variable starting materials from heavily pretreated lymphoma patients. During clinical development of axi-cel, a manufacturing success rate of >99% was achieved in a trial with 101 treated patients.
To successfully complete process performance qualification as part of process validation, and to establish an appropriate control strategy for commercial application, process understanding must be well established. During process characterization, it is possible to identify both critical and noncritical process parameters that help define appropriate process controls. With this understanding, and a clear recognition of variability introduced through raw materials and process equipment, tight control over unit operations is achievable. Although individual unit operations may be well controlled and conducted as closed-system operations, the manufacturing process for axi-cel described here does not rely completely on automated operations. Despite significant improvements, future innovation to remove sources of variability from the process will be extremely valuable. A well-grounded control strategy and established framework for evaluating comparability will ensure that the introduction of any future process improvements can be evaluated objectively for potential impact on process performance.
Author disclosure
All authors are employees of Kite, a Gilead Company with equity ownership.
Acknowledgements
This work would not have been possible without the hard work and dedication of colleagues from Kite. We are particularly thankful to TJ Langer, James Oliver, Jimmy Glenn, Chris Shen, Andrew Dang, Tawny Watanabe, Sana Sirajuddin, Emily Lowe, John Rossi, Allen Xue, and Adrian Bot.
References
1. Kershaw MH, Westwood JA, Parker LL et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006; 12: 6106–6115. CrossRef
2. Lamers CH, Sleijfer S, Vulto AG et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J. Clin. Oncol. 2006; 24: e20–e22. CrossRef
3. Park JR et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol. Ther. 2007; 15: 825–833. CrossRef
4. Till BG, Jensen MC, Wang J et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood 2012; 119: 3940–3950. CrossRef
5. Grupp SA, Kalos M, Barrett D et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013; 368: 1509–1518. CrossRef
6. Kochenderfer JN, Dudley ME, Kassim SH et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015; 33: 540–549. CrossRef
7. Davila ML, Riviere I, Wang X et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014; 6: 224ra25. CrossRef
8. Brentjens RJ, Davila ML, Riviere I et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013; 5: 177ra38. CrossRef
9. Porter DL, Levine BL, Kalos M et al.Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011; 365: 725–733. CrossRef
10. Lee DW, Kochenderfer JN, Stetler-Stevenson M et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015; 385: 517–528. CrossRef
11. Kochenderfer JN, Feldman SA, Zhao Y et al. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J. Immunother. 2009; 32: 689–702. CrossRef
12. Kochenderfer JN, Wilson WH, Janik JE et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010;116: 4099-4102. CrossRef
13. Tumaini B, Lee DW, Lin T et al. Simplifier process for the production of anti-CD19-CAR-engineered T cells. Cytotherapy 2013; 15: 1406–1415. CrossRef
14. vanSchalkwyk MCI, Papa SE, Jeannon JP et al. Design of a Phase I Clinical Trial to Evaluate Intratumoral Delivery of ErbB-Targeted Chimeric Antigen Receptor T-Cells in Locally Advanced or Recurrent Head and Neck Cancer. Hum. Gene Ther. Clin. Dev. 2013; 24: 134-42. CrossRef
15. Better M, Chiruvolu V, Oliver J et al. Manufacturing and characterization of KTE-C19 in a multicenter trial of subjects with refractory aggressive non-Hodgkin Lymphoma (NHL). Cancer Res. 2016; 76(suppl, abstr): 2308.
16. Lu TL, Pugach O, Somerville R et al. A rapid cell expansion process for production of engineered autologous CAR-T cell therapies. Hum. Gene Ther. Methods 2016; 27: 209–218. CrossRef
17. Locke FL, Neelapu S, Bartlett NL et al. Phase 1 results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol. Ther. 2017; 25:285-295. CrossRef
18. Locke FL, Neelapu S, Bartlett NL et al. Primary results from ZUMA-1: a pivotal trial of axicabtagene ciloleucel (axi-cel; KTE-C19) in patients with refractory aggressive non-Hodgkin lymphoma (NHL). Cancer Res. 2017;77(suppl, abstr):CT019.
19. Levine, BL et al. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev 2017; 4: 92–101. CrossRef
20. Locke FL, Rossi J, Xaue X et al. Immune signatures of cytokine release syndrome and neurologic events in a multicenter registrational trial (ZUMA-1) in subjects with refractory Non-Hodgkin lymphoma treated with axicabtagene ciloleucel (KTE-C19). Cancer Res. 2017; 77(suppl, abstr): CT020.
21. US Food and Drug Administration. Guidance for Industry. Process Validation: General Principles and Practices.
22. Kochenderfer JN, Somerville RPT, Lu T et al. Lymphoma remissions caused by anti-CD19 chimeric antigen receptor T cells are associated with high serum interleukin-15 levels. J. Clin. Oncol. 2017; 35: 1803–1813. CrossRef
23. Locke FL, Rossi J, Neelapu SS et al. Product characteristics associated with in vivo expansion of anti-CD19 CAR T cells in patients treated with axicabtagene ciloleucel (axi-cel). J. Clin. Oncol. 2017; 35(suppl, abstr): 3023.
Affiliations
Marc Better1*, Vijay Chiruvolu2 & Marianna Sabatino3
1 VP, Product Sciences.
2 VP, Process Sciences and Engineering.
3 Senior Director, Product Sciences.
Kite Pharma, 2225 Colorado Avenue, Santa Monica, CA 90404 USA.
* Author for correspondence: mbetter@kitepharma.com

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License</