Applications and optimization of cryopreservation technologies to cellular therapeutics
Cell Gene Therapy Insights 2017; 3(5), 359-378.
10.18609/cgti.2017.038
Delivery of cell therapies often requires the ability to hold products in readiness whilst logistical, regulatory and potency considerations are dealt with and recorded. This requires reversibly stopping biological time, a process which is often achieved by cryopreservation. However, cryopreservation itself poses many biological and biophysical challenges to living cells that need to be understood in order to apply the low temperature technologies to their best advantage. This review sets out the history of applied cryopreservation, our current understanding of the various processes involved in storage at cryogenic temperatures, and challenges for robust and reliable uses of cryopreservation within the cell therapy arena.
The explosion of interest in cell therapies over recent years has necessarily focused attention on the processes that will enable product delivery in reliable, regulatory-compliant and robust ways. These considerations sometimes introduce new challenges that may not have been important in the original laboratory studies on that particular cell therapy. One such consideration is product storage (cryobanking), which is often required to sustain cell therapy delivery to the end user facilities at the required time, providing a quality assured product with required safety and potency characteristics [1,2]. Cryopreservation in its various forms is one of the main facilitatory technologies for cell therapies to be able to meet these demands. This brief review will discuss the salient topics within this field and include the history of cryopreservation, what we currently understand about the biophysical and biological processes that allow successful cell recoveries after storage, and additional challenges for scale up and regulatory oversight when the technology is applied.
History of Cryopreservation
The widely accepted term to describe preservation of living cells, capable of biological reanimation, is cryopreservation. In reality, cell biopreservation can be achieved across a range of reduced temperatures, which enhance survival by reducing biological activities, but with limitations dependent upon the chosen modality. The common conception of cryopreservation is storage of living cells at the deep cryogenic temperatures provided by liquid nitrogen or the associated vapour phase (ranging from -196o to approximately -170oC).
The challenge for biology of course is the phase change of water that occurs at the rather inconveniently high temperature (for biopreservation purposes) just below 0oC. There have been many studies over the past few hundred years on the biological effects of freezing, and the allied phenomenon of freeze tolerance in overwintering species (most often in the plant kingdom). The growing understanding of freezing effects followed the development of microscopes capable of directly observing the freezing process; for example Molisch [3] described the freezing process in plant tissues, which immediately highlighted one of the central problems with ice formation – the fact that ice, derived as the phase change of pure water, resulted in exposure of the cells to a residual hypertonic environment as solutes (originally dissolved in the aqueous environment) are excluded from the ice crystal lattice. In simple terms, the cells experienced a lethal osmotic stress that could be detected at the structural level very quickly after thawing. Other scientists of the same era, such as Maximov [4] provided evidence that in plants capable of overwintering in northern Russia, the tissues went through a seasonal hardening process, which was accompanied by accumulation of solutes such as sugars. Chambers and Hale [5] at the turn of the 20th Century provided additional evidence from light microscope studies that there was an osmotic effect of freezing directly on the plant cells that was accompanied by lysis on thawing. Over the subsequent 50 years, others continued to explore the biophysical principles and biological effects of the water–ice phase transition; for example Luyet [6] made many pertinent observations on ice crystal structures, changes in these brought about by alterations in the kinetics of cooling or the presence of solutes in the aqueous medium, and the effects on living cells. This collected knowledge base undoubtedly influenced Polge [7] and his colleagues in their studies in 1949 on freezing of reproductive cells (notably fowl spermatozoa to enhance animal breeding in the period post the Second World War) which resulted in the first clear evidence of recovery of functional cells after deliberate deep cryogenic exposure (in their case to -79oC using solid carbon dioxide – liquid nitrogen was not readily available at that time). The key to their success was the exposure of the sperm to glycerol ahead of the cooling process, which Polge later acknowledged was partly by good fortune, but nevertheless (and with the benefit of hindsight), combined all of those previous studies into one successful outcome. That success also fuelled a global effort to better understand and maximize the opportunities provided in biology and medicine for extended biopreservation, culminating in the definition of the term ‘cryobiology’ to encompass the necessary collaboration between biologists, engineers and physicists to better understand the processes, and the establishment of the International Society for Cryobiology in 1963.
Current understanding of cryopreservation
The water–ice phase transition
As is universally accepted, liquid water is the essential component for almost all biological processes [8], and its removal during the formation of ice poses extreme challenges. There have been many excellent reviews on this topic, but a decade ago Mazur [9] and Muldrew and colleagues [10] provided excellent discussions on the topic with specific relevance to cryobiology. These are beyond the remit of the current discussion, but as a brief summary, water exists in the liquid state in a random but self-associating matrix (on an extremely brief timescale) through interactions of hydrogen bonding, which for biology enables solvation of essential ions and solutes, and structural stability of many macromolecules. During cooling, energy within the system is removed and water molecule self-association leads to longer-lived intermolecular connections that result in ice nuclei. In this process, central water molecules hydrogen bond with four surrounding others that at the point of freezing repeat indefinitely throughout the aqueous milieu to yield hexagonal ice with which we are all familiar. The open lattice nature also results in the often observed slight change in density of ice over liquid water. The stabilisation of the network also results in the well-known release of energy detectable as the latent heat of ice formation. The ice crystal lattice cannot maintain previously accommodated solutes, which become excluded into the residual liquid volume surrounding the growing ice interface, which in turn depresses the freezing point of the residual water so that cells are exposed to progressively higher solutes in a progressively smaller liquid water space. Thus ‘freezing’ is not an instantaneous event in most practical applications, even though it may appear so to the naked eye.
Residual mobile water, all-be-it as a tiny fraction of the original water volume, can be detected down to surprisingly low temperatures [11], contributing to the progressive osmotic stress experienced by the cells. Multiple biological targets for this type of injury have been discussed, including destabilisation of cell membranes, change in the intracellular milieu including pH, chemical and structural changes to organelles and to proteins; it is fair to say that we still do not fully understand all the biological consequences [9,10]. What is clear is that in almost all cases, cell membranes and intracellular macromolecules to some degree hinder the kinetics of the formation of ice crystals, such that ice preferentially initiates and grows in the aqueous external solution (i.e., the supporting culture medium in cell therapies), providing the osmotic driving force for water to leave the intracellular environment and effectively shrinking the cells, which can be observed in real-time by cryomicroscopy [12].
Cryoprotectants: solutes that enhance survival during freezing
Given this understanding of events during freezing, we can begin to understand the relevance of glycerol in Polge’s original successful experiments [7], and the observations that over-wintering species frequently accumulate sugars or other polyols [13]. It became clear that such solutes are structurally effective in hydrogen bonding, which in turn implies that they can interact in reversible ways to hydrogen bond with water molecules. This likewise imparts upon them properties to modulate ice formation on a kinetic basis, such that as cooling progresses, less ice is formed at a given sub-zero temperature, often to a greater degree than would be predicted from classical depression of freezing point effects of included solutes. For example, starting in an isotonic culture medium (effectively close to 0.15M sodium chloride), salt concentrations will reach approximately 3.51 molal in the residual liquid fraction by freezing to –5°C, whereas the presence of 1M cryoprotectant (CPA), such as glycerol, mitigates the rise in salt concentration such that even when freezing to below -30°C, less salt is present [10]. This process is described in cryobiological terms as the colligative effects imparted by CPA. Glycerol was quickly applied to studies on freezing other important cell types, such as red blood cells for transfusion. In this era, Lovelock and Bishop described the freeze-modifying effects of dimtheyl suphoxide [DMSO], which equally acts in a colligative fashion, and has now become perhaps the most widely applied protective solute [14]. However, it should be noted that in both these (and subsequent cases), the CPA solutes need to be applied in concentrations far higher than for other solutes normally present in the cell media solutions, which in itself introduces complex biological challenges.
The identification of these freeze-protecting solutes led by the 1970’s to the term ‘cryoprotectant’ (or CPA) to be assigned to solutes that could enhance cell recovery, and an attempt to describe cryoprotectants in classical terms of pharmacology [17]. A whole range of water-modifying agents were listed as CPA, but by the 1980’s the list of widely used CPA with good efficacy had been refined down to approximately seven agents (Table 1). It became understood that some of the CPA (such as DMSO or glycerol) would cross cell membranes and provide intracellular protection, whilst other agents such as sugars or polymers could provide CPA effects in cell systems where little intracellular permeation was taking place, which has been attributed to their ability to produce partial dehydration and limit harmful intracellular ice formation (see section below). This has led to the pragmatic classification of CPA as either cell-permeating or non-permeating agents [15]. Another important point is that CPA must also remain in solution at very low temperatures during the cooling process – solutes that precipitate out at high subzero temperatures cannot effectively modify ice formation during cryopreservation. Whilst wishing to avoid over-simplification, it is a ‘rule of thumb’ that nearly all nucleated mammalian cells require intracellular protection during cryopreservation; non-permeating CPA can provide additional benefits to modulate ice crystal growth in the extracellular environment (and thereby help mitigate the osmotic effects of ice formation), but they cannot normally provide primary cryoprotection [15]. This statement has to be moderated to some degree when considering cryoprotection afforded by sugars such as trehalose and sucrose, which can be used to entirely replace agents such as DMSO for some cell types; however, in this situation it is clear that the sugars are needed both extra- and intracellularly, which can be achieved by pre-cryopreservation culture for 24 hours [18,19]. Nevertheless, these additional effects of agents such as polymers and proteins should not be discounted when considering how to develop a CPA protocol for a given cell therapy, and can explain to some degree why for laboratory scale cryopreservation, many groups have used high protein concentrations such as foetal calf serum in the freezing media in their cryobanking activities.
| Table 1: Common cryoprotectant (CPA) identified by widespread***, moderate**, or infrequent choice of agent | ||
|---|---|---|
| Cell permeating agents | Sugars (which may permeate cells to a degree depending on molecular size) | Polymers |
| Dimethyl sulphoxide*** | Sucrose*** | Polyethylene glycol (PEG)*** |
| Ethylene glycol*** | Trehalose*** | Hydroxy ethyl starch*** |
| Propylene glycol*** | Raffinose** | Polyvinyl pyrrolidone (PVP)** |
| Glycerol** | Mannitol** | Ficoll** |
| Methanol* | Glucose* | Serum proteins (mixture)** |
| Ethanol* | Galactose* | Milk proteins (mixture)** |
| Particular CPA mixtures are often selected for specific cell preservation strategies. This list is not exhaustive and a wider discussion on CPA can be found in [15,16]. Oligosaccharides tend to act as non-permeating osmotically acting CPA, whereas monosaccharides may permeate mammalian cells to a degree depending on cell type. | ||
It will be clear by now that CPA have important roles as water-modifying agents during freezing stress, but those same properties can cause problems for generalized cell function. Having high concentrations of a particular solute in the intracellular environment will impact on almost all normal biological processes before or after cryopreservation, and this interference can at some point become toxic, leading to cell injury even before ice formation. This is equally true in osmotic terms because, even though permeating CPA may be chosen for use, they are needed in relatively high concentrations and they will take an identifiable time to cross the cell membrane, much slower than for water movement itself. Thus when exposing cells to CPA at concentrations of 0.5M or above, there is an initial osmotic shrinkage of cells as water leaves, to be balanced by a cell volume re-equilibration as CPA and associated water molecules enter the intracellular space [20]. Transmembrane CPA permeation is largely a simple physico-chemical process, the kinetics of which mean that at ambient temperature for example, CPA permeation into mammalian oocytes as a model system requires some 10 minutes to approach equilibrium [21]. The process is generally faster at higher temperatures, for example in mammalian cells cultured at 37oC, but higher temperatures tend to exacerbate any chemical toxicities for the CPA. Low temperatures for CPA exposure (such as 10oC or below) can mitigate chemical toxicity but also prolong exposure time to achieve good intracellular permeation, often by a factor of two or more [22]. Whilst some of these factors can be predictively modelled once variables such as cell membrane permeability coefficients are known for water or CPA [23], there is currently no absolute substitute to performing prospective investigations to define the best CPA protocols to maintain good potency for specific cell therapies during the cryopreservation process. Recently the application of algorithm-based objective optimisation of cryoprotectant protocols is providing valuable information on these important areas for cell therapy cryopreservation [24].
The kinetics of cooling for successful cryopreservation and storage considerations
Successful cryopreservation was also found to be influenced by the kinetics of the cooling process itself. As often practiced today, slow cooling (where slow as a relative term applies to cooling rates of between about -0.3oC min-1 and -2oC min-1) were found by empirical observation to relate to good success. Mazur and colleague provided a hypothesis based on these observations, which he developed over many years [25]. It can be described as Mazur’s two-factor hypothesis and explained visually in Figure 1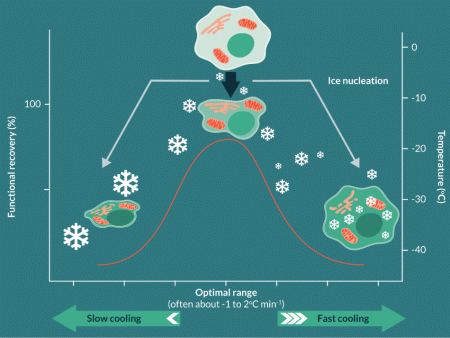
To survive cryopreservation, cells are required to be optimally dehydrated to avoid intracellular ice formation; intracellular freezing was generally found to be associated with lethal cell injury in the vast majority of cases [26]. At an optimal slow cooling rate, cells could be dehydrated with their sensitive molecular and ultrastructural components protected by the added CPA. However, if very slow cooling (around -0.1oC min-1 or less) was instigated, cells were exposed to the extreme ice-related dehydration for intolerably long times, even in the presence of CPA, and again, lethal injury would ensue. This undoubtedly is an oversimplification of the complex biophysical processes occurring during cryopreservation, many of which remain to be fully elucidated, but the two-factor hypothesis does tend to fit the observed outcomes in many cases over the intervening years. This is where the often quoted ‘-1oC min-1 cooling rate’ has its origins; the ‘-1oC’ is not a mystical number, rather it is a reflection of the time required to achieve the optimal cell dehydration for cell survival during the cryopreservation process. It is extremely important to understand that this is the safe cooling kinetic for the biomass itself when cooling cells, not just the cooling profile of the holding chamber, produced by whatever equipment used to control cooling process. Controlled cooling can be provided by a number of cooling technologies based around liquid nitrogen vapour (e.g., Planer PLC; Cryomed™) or electrical Stirling Engine Systems (e.g., Asymptote PLC). In most cases, cooling small volumes in such equipment (such as traditional 1.8 ml cryovials) will allow the vial contents to closely track the changing temperature conditions within the machine. However, clinical scale cell therapies (discussed below) often require cryopreservation at much larger volumes, where it is a much greater challenge to ensure that the biomass cools at appropriately survivable low rates (discussed below) because of the nature of heat and mass transfer in the large volumes.
The question then arises as to what the desired end temperature should be for success, and the requirement for temperature control over that range. It has now been established from a number of studies that for true long-term stability, temperatures below -100oC become essential [27]. Cryobiology has provided information on the physico-chemical status of the frozen matrix which indicates that in the range -120 to -130oC, a glassy transition occurs that finally and completely solidifies the mixed matrix of ice, solutes, CPA and biomass in the samples. This temperature range relates to the total solute concentration of the mixture, but for all practical purposes in cell therapy the quoted figures are relevant. It has been established empirically in different systems however, that practical benefits for slow cooling beyond about -60oC to -80oC are not detectable, so cryopreservation protocols often terminate the imposed control of slow cooling at these temperatures [25,28]. Thereafter, the samples can be cooled below the glass transition to close to the storage temperature in liquid or vapour phase nitrogen (for example to -160oC) at faster, pragmatically convenient, (but still controlled) rates of about -10o or -20oC min-1 [29]. Effectively by about -60oC, the residual solution in the sample, containing now highly concentrated solutes and CPA, becomes so highly viscous that the glass transition event (Tg) becomes highly probable on further cooling. Within this matrix, the biomass is present as extremely dehydrated cells that contain no ice [30]. A schematic of a slow cooling process is provided in Figure 2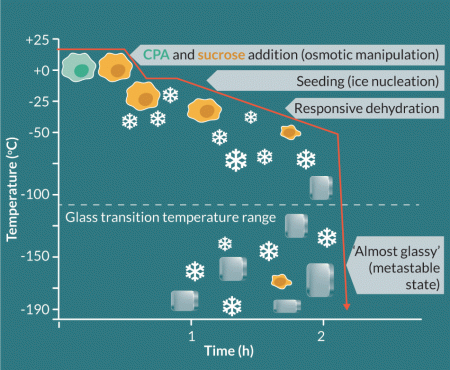
The value of liquid nitrogen as the storage medium of choice will be now be apparent because the temperature provided is far below that of the glass transition temperature range, imparting a degree of safety to the process. The stable cryogenic temperatures (below -150oC) that can be obtained (with modern storage containers fitted with appropriate monitoring and if needed, autofill devices) are sufficiently far enough below the glass transition range to yield true long-term biopreservation on a scale probably greater than required for most cell therapies. It has been often discussed that below Tg and in the highly viscous environment, there is insufficient thermal energy for any chemical reactions or molecular diffusion to take place. The only potential harmful process to impact cryogenic storage has been suggested to be background ionising radiation, and it has been estimated that it would take in excess of 104 years for cells at cryogenic temperatures to accumulate lethal injury [1]. In practical terms, there have been reports of cryopreserved cells, such as sperm or embryos, retaining normal functional processes after up to about 30 years cryo-storage [31,32]. Likely of more relevance to cell therapies, cells from umbilical cord blood have been used effectively after 11 years cryo-storage [33].
Historically, storage at -80oC using electrical freezers has been used for some cells such as red blood cells [34]. However, it will be clear from above discussions that storage above Tg is likely to result in slow but progressive cryo-attrition. In practical terms, a storage shelf-life has often been imposed. The FDA has approved -80oC storage of red blood cells for 10 years [35]. For more complex systems, storage at -80oC for prolonged periods may be more problematic for example, in work on liver cell spheroid cryo-storage it was shown that significant cryo-attrition occurred over 1 year of storage [36]. For each cell therapy, the ease of application for storage at -80oC needs to be balanced by an understanding of product stability. The advent of electrical freezers operating in the -120oC range has opened this possibility as a storage temperature for cell therapies, which is predicted to have successfully long storage times, but little published information on that is currently available for prospective studies comparing these technologies with liquid nitrogen storage.
Given the expected stability of storage below Tg, the occasional report on negative effects of storage duration [37] are difficult to understand on a physico-chemical basis. However, during practical storage of cell products, there is always a potential for fluctuations in storage temperature if cryogen levels are allowed to vary widely (e.g., during intermittent delayed times of filling with liquid nitrogen) or if samples, held in racks or trays, are removed from storage whilst accessing specific products on day of requirement. Even though the products may appear visibly ‘deep frozen’ during this process, it has been shown that significant temperature upshift (e.g. from -135oC to -60oC) could occur within a few minutes of lifting racks out from storage [38]. Repeated temperature cycling through the range for Tg in this fashion led to a decrease in cell functional recoveries.
The choice of storage in liquid or vapour phase nitrogen for cell therapies has trended towards vapour phase storage. The possibility of transfering infectious agents found in commercially produced liquid nitrogen has been discussed and experimentally demonstrated [39]. Vapour phase storage (with temperatures usually in the region of -150o to -170oC) is attractive, but potentially capable of rapid temperature fluxes such as when opening the storage container. Happily, the placement of commonly used aluminium racks within the storage tank acts as a ‘cold sink’ to stabilise the internal environmental temperature, and this can be further stabilised by simple processes such as adding a copper fin [40] within the storage tank. For optimal storage of cell therapies, cryo-bank access procedures should be subject to protocol management and routine data record.
Considerations for warming
For successful recovery of potent cell therapies, the reversal of all the above described biophysical events has to be performed in ways which do not compound injury. For the most part, cell therapies are traditionally subject to cooling, in the presence of significant ice content in the sample. It has long been recognised that during the warming process, existing ice crystals may undergo ice crystal growth and re-organisation (known as Ostwald ripening), which may impart further injury depending upon the kinetics of the process [41]. In addition, if small intracellular ice nuclei were established during the cooling process, but did not grow to become injurious, these may grow during warming, again depending on the kinetics of warming, such that ‘freezing during thawing’ can be conceptualised [42]. For these various reasons, fast warming has traditionally been favored [9,43]. In fact, for slowly cooled cryopreserved cells, the impact of warming kinetics is complex and not easy to predict. It has been alternatively argued that slow warming may be beneficial to allow time for osmotic re-equilibration processes to take place [25] when the highly shrunken cells start to encounter more liquid water as the ice matrix melts at high subzero temperatures. In some experiments on particular cell types, the impact of slow versus fast warming was either not significant or was related to the particular CPA used [44]. This raises another potential warming factor – exposure of the cells to toxic high CPA concentrations at high subzero temperatures during slow warming. Warming is an area which deserves greater future investigation. As in cooling, terms such as ‘fast’ or ‘slow’ warming are relative and should be defined in any cell therapy protocol for process repeatability.
Cryopreservation and cell product potency
In many cases, the potency of thawed cell therapies may not equate to what might be predicted from the doses selected for cryopreservation. It has been known for some time that a series of sublethal stresses can accumulate in the cells during the multiple steps of cryopreservation, and may only be expressed gradually after rewarming in a process known as ‘cryopreservation cap’ or ‘cryopreservation-induced delayed onset cell death’ [45,46]. Cell death pathways such as apoptosis and necrosis have been identified. There appears to be a progression of expressed injury over the first 12–24 h post-thaw, and subsequently surviving cells may resume normal function including cell division [29]. Targeted molecular strategies have been suggested to mitigate against these injuries [47] but for clinical application these need to be regulatory approved strategies. These post-thaw injury markers also offer one way to audit effectiveness of cryopreservation protocols or assess protocol change.
Cryogenic preservation in the absence of ice: vitrification
The concept that biological cryopreservation could be achieved by avoiding ice injury with processes that entirely inhibit crystal growth has been considered for many years; the original paper by Polge and colleagues [47] used the term ‘vitrification’ to denote that something unusual was happening which allowed sperm survival (although their system does not match up with what we understand by ‘vitrification’ today). At around the same time, Luyet and Gehenio made observations of an ice-free solidification of protoplasm which required ultra-rapid cooling (several thousand oC min-1) [6]. Later, Rall and Fay were able to translate the ideas into an achievable technology for successful cryogenic preservation of mouse embryos [48]. Vitrification as currently practiced requires very high concentrations of CPA (as much as six times those used in traditional slow cooling cryopreservation), which cells can only tolerate for short periods (usually seconds of exposure time) before they become toxic, and thus still requiring rapid cooling and warming (which in turn limits the volumes used to [49], and Figure 3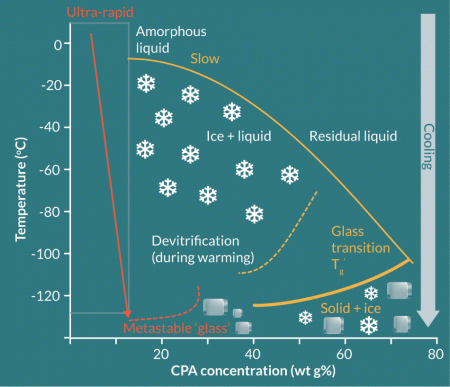
The technology has recently become widely applied in reproductive cryo-banking for infertility treatment [50]. However, because of the volume limitations, vitrification has not yet been developed into technologies for what might be considered more general cell therapy cryo-banking. Recent studies have been made into development of vitrification technologies compatible with large volumes (see Puschmann et al [51] for a review), but these have yet to be applied to deliverable cell therapies.
Applications of cryopreservation for cell therapies & challenges for robust product delivery
Historical and current cell therapy cryopreservation
Cryopreservation of bone marrow-derived hematopoietic progenitor cells (HPC) is an example of a cell therapy product which has been applied widely since the 1960s [52,53]. The principles of slow cooling cryopreservation (around -1oC min -1), with DMSO at 10% w/v as CPA and storage at either -80oC or, less than -130oC have been routinely used [54]. Using secondary CPA such as hydroxyl ethyl starch has allowed reduction of DMSO to 5% concentrations, with hydroxyl ethyl starch added at 6% w/v [55]. Special cryopreservation bags (e.g. Miltenyi Biotec or MacoPharma) with a range of fill volumes up to 250 ml allow maintenance of suitable cooling profiles and good warming rates when immersed in a waterbath at 37oC [56]. Post-thaw manipulation of HPC has often been avoided by direct transfusion of the thawed cell product, but this can occasionally lead to patient-related adverse events [54]. Some authors developed washing procedures to dilute CPA before infusion, as a way to reduce any patient-related adverse events following infusion of DMSO, with acceptable cell product survival [57].
As other cell therapies have entered the arena within the broad area of hematopoietic stem cell replacement, slow cooling cryopreservation has maintained its beneficial role, but with some addition to detail in terms of protocol management. For example, when cryopreserving umbilical cord blood, Woods and colleagues [58] and Hunt et al [59] investigated steps to limit CPA toxicities when applying DMSO. There have been several reports of mesenchymal stromal cells for expansion and cryopreservation for clinical applications, such as in graft versus host disease [60], but the product potency has come into question [61] and cryopreservation was one of the steps under scrutiny. Further detailed studies indicated a range of reversible and non-reversible effects of cryopreservation in mesenchymal stromal cells using slow cooling with a range of DMSO concentrations and cell pre-treatments [62]. Post-thaw culture for 24 h was found to be effective in reparation of many of the cryo-induced abnormalities. A recent review of cryopreservation practices across UK centres applying slow cooling cryopreservation for peripheral blood stem cells identified differences in practices which were reported under the umbrella term ‘cryopreservation’ [63]. These combined studies highlight an important point for all cell therapy cryopreservation activities; this is that cryopreservation is not a simple ‘standardised one option’ process, the various protocol steps need to be well documented in standard operating procedures, and the equipment involved should be applied with care and monitored frequently against documented performance criteria. The authors also cautioned that straightforward assays of cell viability post-thaw may not correlate well with other more exacting tests of product potency, which again is something that needs to be part of protocol testing and audit for any cell therapy cryopreservation practices.
With the recent arrival of manipulated T cell therapies, such as chimeric antigen receptor engineered (CAR ) T cells destined for autologous transfusion [64,65], cryopreservation will undoubtedly remain the important facilitatory technology in the pathways for product management, batch safety and potency validation, and, in many cases, delivery to end users. Cryopreservation has already been discussed in these terms [64] but cryo-protocol details are as yet sparsely reported and it is likely that general slow cooling regimes originally developed for HPC have been adopted. Here also, product depletion and post-thaw potency have been reported [66]. Other manipulated allogeneic immune cell therapies (such as natural killer cell or regulatory T-lymphocyte therapies) face the same requirement for cryopreservation to meet ‘off the shelf’ product delivery, but again post-thaw attrition in cell responses has been highlighted [67]. There is an opportunity for much further work on fundamental cryobiology and translational cryopreservation research in the CAR-T cell arena to improve potency. In the short term, it may be possible to mitigate against loss of potency post-thaw by adjusting dose (delivered cell numbers) but this still leaves difficult questions if the impact of cryopreservation on the therapeutic cell population is not fully understood, and which may therefore vary between batches.
Challenges for scale up for large volume cell therapies
In the discussions so far, cryopreservation has been applied to single cell products at a scale very similar to those used traditionally for haematopoietic stem cells. In other areas, larger cell therapy volumes are required, e.g., cell therapy for organ support such as the bioartificial liver where multicellular units in volumes of between 1–2 litres are required at clinical scale [68]. A potential difference in approach to cryopreservation for multicellular products (such as cell spheroids) under consideration is the impact of controlling ice nucleation during slow cooling. We have discussed the fact that optimal cryo-dehydration is essential for cell survival, which is achieved by the osmotic effects of the growing ice content in the sample. This implies that ice crystal initiation should be a repeatable process during cooling to achieve a standardised process, but ice nucleation is a stochastic event, and on any one cooling run can occur unpredictably at different temperatures far colder than the equilibrium melting temperature of the mixture (where ice would first be able to co-exist with liquid) in different samples [69]. Controlled ice nucleation can be achieved by momentary application of a cold metal rod to the outside of a container such as a cryo-vial in the process known as ‘ice seeding’ and has become routine in reproductive cryopreservation where there are only a small number of samples per run [70]. This is a physical event, whereby local deep cooling applied to the surface of the container produces ice crystals on the inside wall – it has to be visually checked that ice is present internally in the vial and not just ‘frosting’ on the external surface. Ice nucleation can also be induced across a group of vials being slowly cooled together by introducing a brief ‘shock cool’ step with rapid cooling in the cryo-cooler for approximately 2 minutes [71,72] before returning to the programmed slow cooling protocol. Organisation of water molecules into ice nuclei can also be achieved by certain types of physico-chemical surface properties, (often termed ice nucleating agents) for example, crystalline cholesterol, included in the CPA mix, and providing a more reproducible cell cryo-dehydration and often increased cell recoveries [29]. Other types of inorganic ice nucleating surfaces are under development [IceStart™, Asymptote]. Another problem with cryopreservation of large samples is maintaining consistent heat transfer throughout the sample volume for homogenous cooling, since cooling devices work by surface cooling, which depends on the heat transfer properties of the container and the cell mixture [73]. The appropriate placement of thermocouples in sample containers during experimental cryo-cooling runs allows assessment of differences between different sample compartments, or individual samples, and the cooling device chamber [29,73]. Any differences can be mitigated by altering the cooling programme if required. The desired control of cooling can be further disrupted by the release of latent heat of ice nucleation deep within large aqueous volumes [74]. Alterations of the cooling ramp of the cooling machine, such that heat extraction is maximised over a particular but defined range of temperatures where the latent heat effect predominates is one way to improve overall sample cooling profiles [30].
When considering warming rates for larger cryopreserved volumes, similar concerns exist about homogenous heat transfer; the usual process of immersing cryopreserved samples in a warming bath may leave core volumes in a frozen state for extended periods and not achieving the desired rapid warming profile [73,74], thereby exposing cells to CPA at potentially toxic temperatures. Warming is one area of cryopreservation that requires much further study, and understanding the interactions between optimal cooling and warming profiles to maximise cell recovery and potency could benefit from this.
Commercially available containers (cryo-bags) enable cryopreservation of biomass up to volumes of approximately 250 mL and are filled to a recommended volume to produce a sample height of less than 5 mm. This thin envelope configuration ensures that the contents of the bag are cooled homogenously; the relative thinness of the bag also facilitates fast warming rates. However, the dimensions of the current cryobags cannot sustain volumes greater than 250 mL whilst maintaining less than 5 mm height. Increasing the height of the bag compromises the heat transfer process and creates a temperature gradient within the biomass. At volumes greater than 250 ml, maintaining the height at less than 5 mm requires a bag with impracticable dimensions (length and width) to freeze with the current accessible technology (i.e., controlled rate freezers). Therefore, the design of the cryo-container is also a factor under careful consideration when cryopreserving large volume cell therapies, as it must support the homogenous freezing and thawing of the biomass.
Another important key aspect for successful cell therapy is the quick recovery of the biological constructs after cryopreservation in a relevant time frame for immediate availability to the patients. Similar to the production process, recovery could be achieved by transferring these cell constructs into a bioreactor which provides a dynamic environment aiming to mimic the perfusion condition which cells are subjected to in vivo, with more effective mass transfer properties [75]. A number of different types of bioreactors exist (fluidised bed, rotary cell culture system) that can optimally support cell recovery and metabolism whilst minimising cell damage [76-79]. Such systems can be readily scaled-up for fast recovery of large volumes of cryopreserved cell therapies.
Cell encapsulation within hydrogels is widely used in some cell-based therapies, as biocompatible materials can isolate therapeutic cells from the host, avoiding rejection, providing a physical support, and allowing a controlled release of cells from biodegradable materials. The process of encapsulation typically uses liquid precursors that become solid upon polymerization or polymer chain crosslinking. Cryopreservation of encapsulated cells must ensure maintenance of the chemical structure and physical characteristics of the biomaterial as they greatly influence cell behavior and may potentially affect the outcome of the therapy. Several studies have shown promising protective effects of biomaterials on cell cryopreservation [80]. In a more mechanistic study, alginate hydrogel was shown to inhibit devitrification during the warming process of vitrified stem cells [81]. Altogether, these beneficial properties make encapsulation an encouraging strategy for cell therapy and the cryopreservation process.
Considerations for GMP transfer of cell therapy cryopreservation technologies
Moving cell therapy cryopreservation to clinical application will predictably require maintenance of the same GMP-aligned steps that are important across the whole cycle for product delivery. All pre- and post-cryopreservation product manipulations must be performed in ways that adhere to regulatory requirements, using media formulations, CPA additives, containers (such as cryo-bags), cryo-cooling equipment, identifying labelling and traceability documentation, storage temperature monitoring and product delivery systems; all of equal importance [1]. The value of producing a comprehensive quality programme for a cryopreserved cell therapy (in this case peripheral blood mononuclear cells) has been discussed by Ducar and colleagues [82]. DMSO has so far been the most widely used CPA, which must be of highest quality and endotoxin-free. One point of discussion has been the use of regulatory-compliant protein sources (such as foetal bovine serum or human serum) [83] or xeno-protein free solutions [84] as a CPA carrier media. Some commercially available GMP-compliant CPA solutions have recently become available (CryoStor™, BioLife) [2]. For specific types of non-autologous cell therapies, the importance of recording and testing of cell provenance throughout the development of master and working cell banks is an additional important consideration [85].
A number of options are available for cooling equipment when applying controlled slow rate cryopreservation. Passive cooling devices may be suitable for small volumes and sample numbers of a particular product type, such as Mr Frosty™ (Thermo Fisher Scientific) or CoolCell®, (Biocision). Equipment capable of handling larger volumes or more exacting cooling profiles are available based on liquid nitrogen technology (Planer PLC; Cryomed™) or electrical Stirling engine principles (Asymptote Ltd). The importance of understanding the true sample cooling profile (which can be audited by a recording system in a dummy vial) as opposed to the selected chamber program has already been discussed above [29,74]. For storage, filling and cryogenic transportation of cell therapy products, a number of companies provide a range of regulatory compliant options [Panasonic; Thermofisher Scientific; Praxair; Thames Cryogenic]. Liquid nitrogen is not a sterile product unless specifically treated, and thus protocols for handling and storage need to be developed that avoid compromising GMP environments [39,86]. Since nitrogen vapour can deplete room oxygen levels, air oxygen monitors are required where the liquid nitrogen is located and handled as part of general risk assessments and safety measures, which also include personal safety equipment to avoid ‘cold injury’ from accidental contact with the cryogen [87].
Technical innovations continue to come on stream to make cryopreservation more robust in the context of regulatory oversight for delivering cryo-banked cell therapies. Labelling systems that survive cryogenic exposure are provided by a range of companies (GA International; Biosafe). Thawing devices capable of imposing and recording warming profiles for cryopreserved product in either cryo-vial or cryo-bag formats are now available (Medcision™; Asymptote). Secure closure vials and automatic fill systems can meet the demands for some cell therapies (e.g. Cook Regentec; Wheaton®). Lyophilisation (freeze drying) with ambient storage is being developed as an additional biopreservation strategy for some nucleated cell products (Lyotechnology, Osiris Therapeutics Inc.). A number of companies are developing sophisticated cryo-product management systems and cloud-based information repositories that will help drive up standards of best practice and information sharing between organisations. Much information in these areas of cryopreservation is available via specialist academic societies such as the International Society for Cryobiology and International Society for Stem Cell Therapy.
Translational insight
Cryopreservation of cell therapies is an important part of product delivery and process management. When optimally applied, it can also reduce costs by avoiding wastage of product which would otherwise be ‘out of shelf life’. However, there remains a significant opportunity to improve application of the cryopreservation process itself by continued research into both fundamental and applied cryobiology. As different, novel, or large-scale cell therapies come on stream, they will likely require improved approaches to cryogenic preservation.
FINANCIAL & COMPETING INTERESTS DISCLOSURE
The authors have no relevant financial involvement with an organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock options or ownership, expert testimony, grants or patents received or pending, or royalties. No writing assistance was utilized in the production of this manuscript.
References
1. Woods EJ, Thirumala S, Badhe-Buchanan SS, Clarke D, Mathew AJ. off the shelf cellular therapeutics: factors to consider during cryopreservation and storage of human cells for clinical use. Cytotherapy 2016; 18(6): 697–711.
CrossRef
2. Baust JM, Corwin WL, VanBuskirk R, Baust JG. biobanking: the future of cell preservation strategies. Biobanking in the 21st Century. 2015; 864: 37–53.
CrossRef
3. Molisch H. [Untersuchungen über das Erfrieren der Pflanzen]. Science 1897; 6(157): 1002–1003.
CrossRef
4. Maximov NA. Chemical protective agents of plants against freezing injury concerning the nature of the protective effect. Berichte Deutschen Bot. Geselschafft 1912; 30: 504–16.
5. Chambers R & Hale HP. The formation of ice in protoplasm. Proc. Royal Soc. London Series B – Containing Papers of a Biological Character 1932; 110(767): 336–352.
CrossRef
6. Luyet BJ & Gehenio PM. thermoelectric recording of ice formation and of vitrification during ultra-rapid cooling of protoplasm. Federation Proceedings 1947; 6(1): 157.
7. Polge C, Smith AU, Parkes AS. revival of spermatozoa after vitrification and dehydration at low temperatures. Nature 1949; 164(4172): 666.
CrossRef
8. Chaplin M. opinion – do we underestimate the importance of water in cell biology? Nat. Rev. Mol. Cell Biol. 2006; 7(11): 861–6.
CrossRef
9. Mazur P. Principles of Cryobiology. In: Fuller BJ, Lane N, Benson EE (Eds). Life in the Frozen State 2004; CRC Press: Boca Raton FL: 3–65.
CrossRef
10. Muldrew K, Elliott J, McGann L. The water to ice transition: implications for living cells. In: Fuller BJ, Lane N, Benson EE (Eds). Life in the Frozen State 2004, CRC Press. 67–108.
CrossRef
11. Beall PT. States of water in biological-systems. Cryobiology 1983; 20(3): 324–34.
CrossRef
12. Cravalho EG and Diller KR. A cryomicroscope for study of freezing and thawing processes in biological cells. Cryobiology 1970; 6(6): 575
CrossRef
13. Storey KB, Baust JG, Buescher P. Determination of water bound by soluble subcellular components during low-temperature acclimation in the gall fly larva, Eurosta solidagensis. Cryobiology 1981; 18(3): 315–321.
CrossRef
14. Lovelock JE, Bishop MWH. Prevention of freezing damage to living cells by dimethyl sulphoxide. Nature 1959; 183(4672): 1394–5.
CrossRef
15. Fuller BJ. Cryoprotectants: The essential antifreezes to protect life in the frozen state. Cryoletters 2004; 25(6) 375–88.
Webiste
16. Best BP. Cryoprotectant toxicity: facts, issues, and questions. Rejuvenation Research 2015; 18(5): 422–36.
CrossRef
17. Karow AM. Cryoprotectants – a new class of drugs. J. Pharmacy Pharmacol. 1969; 21(4): 209
CrossRef
18. Motta JPR, Paraguassú-Braga FH, Bouzas LF, Porto LC. Evaluation of intracellular and extracellular trehalose as a cryoprotectant of stem cells obtained from umbilical cord blood. Cryobiology 2014; 68(3): 343–8.
CrossRef
19. Rogulska O, Petrenko Y, Petrenko A. DMSO-free cryopreservation of adipose-derived mesenchymal stromal cells: expansion medium affects post-thaw survival. Cytotechnology 2017; 69(2): 265–76.
CrossRef
20. Jackowski S, Leibo SP, Mazur P. Glycerol permeabilities of fertilized and unfertilized mouse ova. J. Experimental Zoology 1980; 212(3): 329–341.
CrossRef
21. Paynter SJ, Cooper A, Gregory L, Fuller BJ, Shaw RW. Permeability characteristics of human oocytes in the presence of the cryoprotectant dimethylsulphoxide. Human Reproduction 1999; 14(9): 2338–42.
CrossRef
22. Paynter SJ, O’Neil L, Fuller BJ, Shaw RJ. Membrane permeability of human oocytes in the presence of the cryoprotectant propane-1,2-diol. Fertil. Steril. 2001; 75(3): 532–38.
CrossRef
23. Paynter SJ, McGrath JJ, Fuller BJ, Shaw RW. A method for differentiating non-unique estimates of membrane transport properties: Mature mouse oocytes exposed to glycerol. Cryobiology 1999; 39(3): 205–214.
CrossRef
24. Pollock K, Samsonraj RM, Dudakovic A et al. improved post-thaw function and epigenetic changes in mesenchymal stromal cells cryopreserved using multicomponent osmolyte solutions. Stem Cells and Development 2017. (EPub ahead of print)
DOI
25. Whittingham DG, Mazur P, Leibo SP. Survival of mouse embryos frozen to -196 degrees and -269 degrees C. Science, 1972; 178(4059): 411
CrossRef
26. Leibo SP, Mcgrath JJ, Cravalho EG. Microscopic observation of intracellular ice formation in unfertilized mouse ova as a function of cooling rate. Cryobiology 1978; 15(3): 257–271.
CrossRef
27. Spurr EE, Wiggins NE, Marsden KA, Lowenthal RM, Ragg Sj. Cryopreserved human haematopoietic stem cells retain engraftment potential after extended (5-14 years) cryostorage. Cryobiology 2002. 44(3): 210–217.
CrossRef
28. Linch DC, Knott LJ, Patterson KG, Cowan DA, Harper PG. Bone-marrow processing and cryopreservation. J. Clin. Pathol. 1982; 35(2): 86–190.
CrossRef
29. Massie I, Selden C, Hodgson H, Fuller B. Cryopreservation of encapsulated liver spheroids for a bioartificial liver: reducing latent cryoinjury using an ice nucleating agent. Tissue Eng Part C Methods 2011; 17(7): 765–74.
CrossRef
30. Massie I, Selden C, Hodgson H, Fuller B, Gibbons S, Morris GJ. GMP cryopreservation of large volumes of cells for regenerative medicine: active control of the freezing process. Tissue Engineering Part C-Methods 2014; 20(9): 693–702.
CrossRef
31. Dowling-Lacey D, Jones E, Bocca S, Stadtmauer L, Gibbons W, Oehninger S. Two singleton live births after the transfer of cryopreserved-thawed day-3 embryos following an unstimulated in-vitro oocyte maturation cycle. Reprod. Biomed. Online 2010; 20(3): 387–90.
CrossRef
32. Feldschuh J, Brassel J, Durso N, Levine A. Successful sperm storage for 28 years. Fertil. Steril. 2005; 84(4): 1017.
CrossRef
33. Mitchell R, Wagner JE, Brunstein CG et al. Impact of long-term cryopreservation on single umbilical cord blood transplantation outcomes. Biol. Blood Marrow Transplantation 2015; 21(1): 50–54.
CrossRef
34. Meryman HT. Red-cell freezing by American-National-Red-Cross. Am. J. Med. Tech. 1975; 41(7): 265–82.
Webiste
35. Scott KL, Lecak J, Acker JP. Biopreservation of red blood cells: past, present, and future. Transfus. Med. Rev. 2005. 19(2): 127142.
CrossRef
36. Massie I, Selden C, Hodgson H, Fuller B. Storage Temperatures for Cold-Chain Delivery in Cell Therapy: A study of alginate-encapsulated liver cell spheroids stored at-80 degrees C or-170 degrees C for up to 1 year. Tissue Eng. Part C – Methods 2013; 19(3): 189–95.
CrossRef
37. Desrosiers P, Legare C, Leclerc P, Sullivan R. Membranous and structural damage that occur during cryopreservation of human sperm may be time-related events. Fertil. Steril. 2006; 85(6): 1744–52.
CrossRef
38. Germann A, Oh YJ, Schmidt T, Schon U, Zimmermann H, von Briesen H. Temperature fluctuations during deep temperature cryopreservation reduce PBMC recovery, viability and T-cell function. Cryobiology 2013; 67(2) 193–200.
CrossRef
39. Grout BWW, Morris GJ. Contaminated liquid nitrogen vapour as a risk factor in pathogen transfer. Theriogenology 2009; 71(7): 1079–82.
CrossRef
40. Hunt CJ & Pegg DE. Improved temperature stability in gas-phase nitrogen refrigerators: use of a copper heat shunt. Cryobiology 1996; 33(5): 544–51.
CrossRef
41. Karlsson JOM & Toner M. Long-term storage of tissues by cryopreservation: Critical issues. Biomaterials 1996; 17(3): 243–56.
CrossRef
42. Rall WF & Polge C. Effect of warming rate on mouse embryos frozen and thawed in glycerol. J. Reprod. Fertil. 1984; 70(1): 285.
CrossRef
43. Baust JM, Corwin W, Snyder KK, Van Buskirk R, Baust JG. Cryopreservation: Evolution of Molecular Based Strategies. Adv. Exp. Med. Biol. 2016; 951: 13–29.
CrossRef
44. Dong QX, Hill D, VandeVoort CA. Interactions among pre-cooling, cryoprotectant, cooling, and thawing for sperm cryopreservation in rhesus monkeys. Cryobiology 2009; 59(3): 268–74.
CrossRef
45. Baust JM, Van Buskirk R, Baust JG. Modulation of the cryopreservation cap: elevated survival with reduced dimethyl sulfoxide concentration. Cryobiology 2002. 45(2): 97–108.
CrossRef
46. Bissoyi A, Nayak B, Pramanik K, Sarangi SK. Targeting cryopreservation-induced cell death: a review. Biopreserv. Biobank. 2014; 12(1): 23–34.
CrossRef
47. Baust JG, Snyder KK, Van Buskirk R, Baust JM. Integrating molecular control to improve cryopreservation outcome. Biopreserv. Biobank. 2017; 15(2):134–141.
CrossRef
48. Rall WF & Fahy GM. Ice-free cryopreservation of mouse embryos at -196-Degrees-C by vitrification. Nature 1985; 313(6003): 573–5.
CrossRef
49. Fahy GM & Wowk B. Principles of cryopreservation by vitrification. Cryopreservation and Freeze-Drying Protocols. 3rd Edition, 2015; 1257: 21–82.
CrossRef
50. Vajta G, Rienzi L, Ubaldi FM. Open versus closed systems for vitrification of human oocytes and embryos. Reproductive Biomedicine Online 2015; 30(4): 325–33.
CrossRef
51. Puschmann E, Selden C, Butler S, Fuller B. Liquidus tracking: controlled rate vitrification for the cryopreservation of larger volumes and tissues. Cryo. Letters 2014; 35(4): 345–55.
Webiste
52. Pegg DE. Freezing of Bone Marrow for Clinical Use. Cryobiology 1964; 51: 64–71.
CrossRef
53. Appelbaum FR, Herzig G, Graw RG, Zeigler JL. accelerated hematopoietic recovery following the infusion of cryopreserved autologous bone-marrow in humans. Experimental Hematol. 1979; 7: 297-–301.
Webiste
54. Pamphilon D. & Mijovic A. Storage of hemopoietic stem cells. Asian J. Transfus. Sci. 2007; 1(2): 71–6.
CrossRef
55. Sputtek A, Jetter S, Hummel K, Kuhnl P. Cryopreservation of peripheral blood progenitor cells: characteristics of suitable techniques. Transfusions Medizin 1997; 34: 79–83.
Webiste
56. Dijkstra-Tiekstra MJ, Hazelaar S, Gkoumassi E, Weggemans E, de Wildt-Eggen J. Comparison of cryopreservation bags for hematopoietic progenitor cells using a WBC-enriched product. Transfus. Apher. Sci. 2015; 52(2): 187–93.
CrossRef
57. Thirumala S, Goebel WS, Woods EJ. Clinical grade adult stem cell banking. Organogenesis 2009; 5(3): 143–54.
CrossRef
58. Woods EJ, Pollok KE, Byers MA et al. Cord blood stem cell cryopreservation. Transfusion Med. Hemother. 2007; 34(4): 276–85.
CrossRef
59. Hunt CJ, Pegg DE, Armitage SE. Optimising cryopreservation protocols for haematopoietic progenitor cells: A methodological approach for umbilical cord blood. Cryoletters 2006: 27(2); 73–83.
Webiste
60. Maziarz RT. Mesenchymal stromal cells: potential roles in graft-versus-host disease prophylaxis and treatment. Transfusion 2016. 56(4): S9–S14.
CrossRef
61. Francois M, Copland IB, Yuan SL, Romieu-Mourez R, Waller EK, Galipeau J. Cryopreserved mesenchymal stromal cells display impaired immunosuppressive properties as a result of heat-shock response and impaired interferon-gamma licensing. Cytotherapy 2012; 14(2): 147–52.
CrossRef
62. Marquez-Curtis LA, Janowska-Wieczorek A, McGann LE, Elliott JAW. Mesenchymal stromal cells derived from various tissues: biological, clinical and cryopreservation aspects. Cryobiology 2015; 71(2): 181–97.
CrossRef
63. Morgenstern DA, Ahsan G, Brocklesby M et al. Post-thaw viability of cryopreserved peripheral blood stem cells (PBSC) does not guarantee functional activity: important implications for quality assurance of stem cell transplant programmes. Br. J. Haematol. 2016; 174(6): 942–51.
CrossRef
64. Levine BL. Performance-enhancing drugs: design and production of redirected chimeric antigen receptor (CAR) T cells. Cancer Gene Therapy 2015; 22(2), 79 –84.
CrossRef
65. Lim WA & June CH. The principles of engineering immune cells to treat cancer. Cell 2017; 168(4): 724–40.
CrossRef
66. Krug C, Wiesinger M, Abken H et al. A GMP-compliant protocol to expand and transfect cancer patient T cells with mRNA encoding a tumor-specific chimeric antigen receptor. Cancer Immunology Immunotherapy 2014; 63(10): 999–1008.
CrossRef
67. Klingemann H. Challenges of cancer therapy with natural killer cells. Cytotherapy 2015; 17(3): 245–249.
CrossRef
68. Selden C, Spearman CW, Kahn D et al. Evaluation of Encapsulated Liver Cell Spheroids in a Fluidised-Bed Bioartificial Liver for Treatment of Ischaemic Acute Liver Failure in Pigs in a Translational Setting. Plos One 2013; 8(12).
CrossRef
69. Morris GJ & Acton E. Controlled ice nucleation in cryopreservation – a review. Cryobiology 2013; 66(2): 85–92.
CrossRef
70. Fuller B & Paynter S. Fundamentals of cryobiology in reproductive medicine. Reprod. Biomed. Online, 2004; 9(6): 680–91.
CrossRef
71. Diener B, Utesch D, Beer N, Dürk H, Oesch F. A Method for the Cryopreservation of Liver Parenchymal Cells for Studies of Xenobiotics. Cryobiology 1993; 30(2): 116–127.
CrossRef
72. Massie I, Selden C, Morris J, Hodgson H, Fuller B. Cryopreservation of encapsulated liver spheroids using a cryogen-free cooler: high functional recovery using a multi-step cooling profile. Cryoletters 2011; 32(2): 158–65.
Webiste
73. Kilbride P, Gonzalez-Molina J, Maurmann N et al. Impact of storage at -80 degrees c on encapsulated liver spheroids after liquid nitrogen storage. Bioresearch Open Access 2016; 5(1): 146–54.
CrossRef
74. Kilbride P, Lamb S, Milne S et al. Spatial considerations during cryopreservation of a large volume sample. Cryobiology 2016; 73(1): 47–54.
CrossRef
75. Martin I, Wendt D, Heberer M. The role of bioreactors in tissue engineering. Trends Biotechnol. 2004; 22(2): 80–6.
CrossRef
76. Hu W, Berdugo C, Chalmers JJ. The potential of hydrodynamic damage to animal cells of industrial relevance: current understanding. Cytotechnology 2011; 63(5): 445–460.
CrossRef
77. Lu J, Zhang X, Li J et al. A new fluidized bed bioreactor based on diversion-type microcapsule suspension for bioartificial liver systems. PLoS ONE 2016; 11(2): e0147376.
CrossRef
78. Serra M, Correia C, Malpique R et al. Microencapsulation technology: a powerful tool for integrating expansion and cryopreservation of human embryonic stem cells. PLoS ONE, 2011; 6(8): e23212.
CrossRef
79. Salehi-Nik N, Amoabediny G, Pouran B et al. Engineering Parameters in Bioreactor Design: A Critical Aspect in Tissue Engineering. BioMed. Res. Intl 2013; 15.
DOI
80. Stéphenne X, Najimi M, Sokal EM. Hepatocyte cryopreservation: is it time to change the strategy? World J. Gastroenterol. 2010; 16(1): 1–14.
Webiste
81. Huang H, Choi JK, Rao W et al. Alginate Hydrogel Microencapsulation Inhibits Devitrification and Enables Large-Volume Low-CPA Cell Vitrification. Advanced Functional Materials 2015; 25(44): 6839–6850.
CrossRef
82. Ducar C, Smith D, Pinzon C et al. Benefits of a comprehensive quality program for cryopreserved PBMC covering 28 clinical trials sites utilizing an integrated, analytical web-based portal. J. Immunol. Methods 2014; 409: 9–20.
CrossRef
83. Vianna LM, Li HD, Holiman JD, Stoeger C, Belfort R, Jun AS. Characterization of cryopreserved primary human corneal endothelial cells cultured in human serum-supplemented media. Arquivos Brasileiros De Oftalmologia 2016; 79(1): 37–41.
CrossRef
84. Miki T, Wong W, Zhou E, Gonzalez A, Garcia I, Grubbs BH. Biological impact of xeno-free chemically defined cryopreservation medium on amniotic epithelial cells. Stem Cell Research & Therapy 2016; 7: 8
CrossRef
85. Stacey G. Banking stem cells for research and clinical applications. Functional Neural Transplantation. Primary and Stem Cell Therapies for Brain Repair, Pt I, 2012; 200: 41–58.
DOI
86. Bielanski A. Biosafety in embryos and semen cryopreservation, storage, management and transport. Reproductive Sciences in Animal Conservation: Progress and Prospects. 2014; 753: 429–465.
CrossRef
87. Day JG & Stacey GN. Biobanking. Mol. Biotechnol. 2008; 40(2): 202–213.
CrossRef
Affiliations
Barry Fuller1*, Jordi Gonzalez-Molina2, Eloy Erro2, Joana De Mendonca2, Sheri Chalmers2, Maooz Awan2, Aurore Poirier2, Clare Selden2
1Division of Surgery & Interventional Sciences
2UCL Institute for Liver and Digestive Health
UCL, UCL Medical School, Royal Free Campus, London NW3 2QG, UK
*Author for Correspondence:
Barry Fuller
Division of Surgery & Interventional Science, UCL Medical School, London NW3 2QG, UK
Phone: +44 207 472 6111
Email: b.fuller@ucl.ac.uk

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.