Non-viral gene transfer with Sleeping Beauty system to engineer T cells for hematologic malignancies and solid tumors
Cell Gene Therapy Insights 2017; 3(2), 301-311.
10.18609/cgti.2017.017
The manipulation of genes has progressed to human application thanks in part to viral-based vectors that transduce a therapeutic gene and sustain its long-term expression. The adoptive transfer of genetically modified T cells against cancer highlights the potential for gene therapy to cure human disease refractory to conventional therapy. The improvements in the genetic manipulation of clinical-grade cells has widened possible treatment options with the realization that personalized gene therapy will be needed in which the introduced gene matches the needs of a particular patient. To match this growing need for individualized therapeutic genes, we have developed a rapid, robust and low-cost approach for clinical translation. This is based on a DNA plasmid-based transposon/transposase, termed the Sleeping Beauty (SB) system. The SB system stably introduces therapeutic genes, such as chimeric antigen receptors (CARs), into clinical-grade T cells and allows for long-term expression of the introduced transgene. Our first-in-human clinical trial of SB-mediated CAR-expressing T cells targeting B-cell malignancies established the safety, feasibility and efficacy of the SB system. This clinical application of the SB system is the foundation for future use of this tool to personalize genetically modified T cells targeting hematologic malignancies and especially solid tumors.
Much progress has been achieved since the first gene therapy trial in 1990 that treated patients with adenosine deaminase (ADA) deficiency using a retrovirus vector to introduce a normal ADA gene into T cells [1]. Retroviral vectors have been the prime tool to introduce therapeutic genes into T cells and hematopoietic stem cells (HSCs). However, the occurrence of deadly leukemia in patients who received cells modified with retrovirus raised concerns about the use of retrovirus, especially in CD34+ HSCs [2,3]. The risk of genotoxicity caused by retroviral vectors seems much less likely in T cells, and this viral vector remains an efficient and useful tool in genetically modified T cell therapy. Another type of viral vector, lentivirus, is also widely used in T cell-based gene therapy in the clinical setting and showed promising clinical results and long-term expression of transgene [4]. Although those viral vector strategies worked well in the early clinical trials, the cost, simplicity, scalability, customization and time to generate clinically compatible viral particles are important issues for future gene therapy using T cells. Naked DNA-based gene therapy might overcome these issues, but it has not been a prime tool for gene therapy until now due to its lower efficiency.
Recent advances in the transposon/transposase system to introduce a therapeutic gene into mammalian cells have the potential to change the landscape of gene therapy. Indeed, we have performed a first-in-human clinical trial using Sleeping Beauty (SB) to introduce a chimeric antigen receptor (CAR) gene into T cells [5]. This trial showed the feasibility and safety of SB-mediated genetic modification of T cells. Other transposon/transposase system-based gene therapy trials are also planned for the near future. The tremendous success of CAR T cell therapy will lead to increasing numbers of clinical trials targeting a variety of different antigens using both CAR and T cell receptor (TCR) [6]. These upcoming trials of T cell gene therapy will rely on the timely and low-cost generation of therapeutic constructs. In this respect, we believe that transposon/transposase-based gene therapy is promising for future clinical application of adoptive cell therapy using genetically modified T cells. In this review, we will briefly summarize the history of gene therapy and the development of the transposon/transposase system, especially the SB system, and discuss the future clinical utility of the SB system in adoptive immunotherapy using genetically modified T cells.
Initial attempts to introduce exogenous genes into human cells
Introduction of foreign genes into human cells can be used to restore or enhance cellular function in human diseases. To sustain the expression of foreign genes, genetic information needs to integrate into the host genome, as integrated foreign genetic information will remain in the host genome even after cellular division. The integration of foreign genes from naked DNA is possible, but the efficiency is extremely low, and usually extensive selection will be required to isolate the cells that have a foreign gene incorporated within the genome [7]. Retroviral vectors can infect mammalian cells through surface receptors, and once they penetrate into the cell they use reverse transcriptase to translate their own RNA into DNA. This DNA is then integrated into the host genome by the virus’s integrase [8]. This process is highly efficient, and by taking advantage of this mechanism, retroviral vectors have been generated by eliminating the replication potential of the native retrovirus. Since the generation of retrovirus vectors, the transduction efficiency of foreign genes has been much improved, especially in dividing cells, and retroviral vectors have become the gold standard tools for gene therapy.
One issue of retroviral vectors is their tendency to integrate into strong enhancers or the promoter regions of active genes [9]. Indeed, the occurrence of acute T-cell leukemia has been reported in patients with X-chromosome linked severe combined immunodeficiency (SCID) who have been treated with HSC genetically modified by retroviral vectors, due to the integration of the retroviral construct into proto-oncogenes, including LMO, and the retrovirus’s long terminal repeat (LTR) enhancer driving their expression [2,10]. Lentiviral vectors may have a safer profile than retroviral vectors [11,12] and are currently under investigation in a clinical trial of genetically modified HSCs [13]. In contrast with the HSC trial, retroviral vectors have never caused leukemia in patients treated with retrovirus-modified T cells. This distinction is likely due not only to the difference in integration sites (active gene in T cell vs HSC) but also the nature of target host cells, as differentiated T cells likely will not cause T cell leukemia even if LMO expression is driven by a strong promoter, as shown in a mouse model [14].
Currently both retroviral and lentiviral vectors are prime tools to generate CD19-specific CAR T cells, and those clinical trials have been highly successful [15-20]. This success increases the enthusiasm to apply CAR T cell technologies to another type of cancer by targeting other tumor associated antigens [21]. Following the initial success of CD19-targeted CAR T cell therapy, many trials are either underway or are planned, targeting many antigens. To match the pace of the immediate needs for clinically compatible gene constructs while minimizing cost will be the next issue to broaden the application of CAR T cell therapy. The cost of CAR T cell therapy is estimated around $500,000 [22]. This is largely due to multiple steps for production of the viral vector and complex verification steps to ensure the viral titer and safety (Figure 1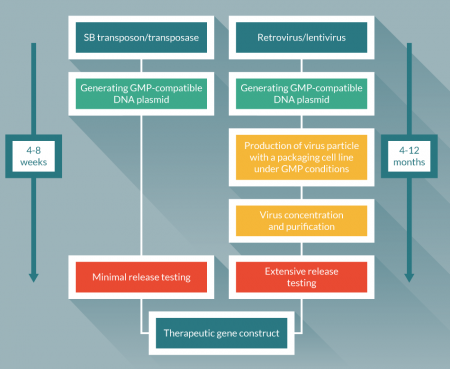
Transposon/transposase
The discovery of jumping genes and the mechanism for this function have led researchers to exploit this system as a valuable genetic tool for mutagenesis and transgenesis [24,25]. From the late 1990’s, several transposon/transposase systems have been found in fish or insects, and those systems has been modified as transgenesis tools for mammalian cells. This development includes the Tol2 [26], piggyBac [27] and SB transposon/transposase systems [28]. Those systems have been shown to efficiently introduce CAR transgene into T cells [29-31].
SB: copy number of integrants
SB was molecularly reconstructed from one of the Tc1/mariner superfamily of transposons isolated from fish. The SB system consists of two components; the first is a transposon that usually carries a gene of interest under a strong eukaryotic promoter, flanked by inverted repeats. The other component, the transposase, was molecularly reconstructed from the Tc-1-like elements from the Atlantic salmon. This has resulted in the SB10 transposase, which possess restored integration activity [28]. This transposase recognizes the inverted repeat sequence and inserts a gene of interest into the host genome by a cut and paste mechanism (Figure 2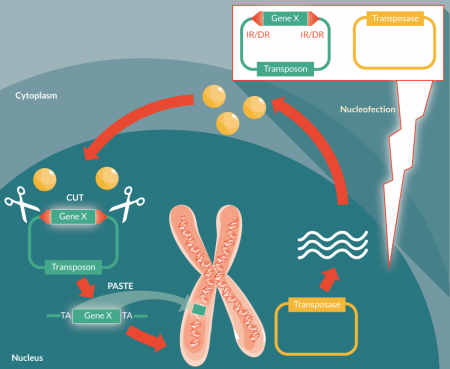
Since the transposase concentration determines the transposition (if the concentration is too high, transposition efficiency will be decreased due to overproduction inhibition), optimization of the transposon to transposase ratio is needed [33]. In our system, using a transposon plasmid to transposase plasmid, we found the ratio of 3 to 1 provides the best transposition efficiency [34]. A further improved, hyperactive SB100x also has been tested in T cells and was found to have enhanced transposition efficiency [35]. SB100x also has been tested in HSCs [36,37] and pluripotent stem cells [38] and showed stable expression of the transgene. The transposition efficiency of CAR construct in T cells may be further improved by the use of a minicircle DNA vector, as recently reported [39].
In our first in human clinical trial, our protocol consisted of repeated stimulation of SB-modified CAR T cells with antigen-specific stimulation by activating and propagating cells (AaPCs) to enrich CAR-expressing T cells over 4 weeks. Interestingly, in our analysis of CAR T cells after four stimulations with AaPCs, the average of integration copy number is about 1.0 per cell, which we think is favorable for gene therapy, as we can minimize the genotoxicity while maintaining expression of the introduced gene [34]. This is opposed to the observation of over 20 copies per cell in some other reports [40]. This discrepancy is presumably due to the difference of transduction (protocol, transposases or size of transgene) and culture protocol of both reports. Repeated AaPC stimulation in antigen specific manner (vs non-specific OKT3 mediated stimulation) may only enrich CAR T cells with low transgene integration.
SB integration sites
The SB system preferentially integrates at TA dinucleotides sites. This preference might also provide an advantage over viral vectors, as the promoter regions in the human genome are known to be GC rich, while TA sites are enriched in introns. From our integration site analysis using ligation mediated PCR and next-generation sequencing, 99.9% of unique integration sites were found at TA dinucleotide sites as expected, and within those 96.5% were in introns [5]. Others have also showed that SB-mediated integration was random and independent of chromatin status (compared to target site distribution of other transposon) [41]. Analysis of CD19 CAR T cells generated by minicircle DNA vectors showed a relatively safer insertion profile compared to lentivirus.
The promise & limitation of SB
One caveat of using the SB system is that its transposition efficiency declines in proportion to the increase in cargo size. When the transgene size is over 4 kb, the efficiency of SB mediated transposition drops significantly [42]. The modified vector platform [43] or the modified inverted repeat structure (sandwich transposon) [44], has been shown to permit larger cargo size gene transduction. Moreover, this may not be an important factor, as most therapeutic genes currently used in the clinic (e.g., CAR and TCR) are less than 2 kb. Thus this issue might not limit the clinical utility of the SB system. Another caveat may come from the transduction method and efficiency. To introduce DNA plasmid into T cells, electroporation is generally used. Due to the large amount of DNA plasmid, cell viability after electroporation is low. The strategy to use smaller size SB constructs (e.g. pFAR4 [45], minicircle vector [39,46]) may improve viability after electroporation and moreover improve transduction efficiency.
First-in-human clinical trials using cells genetically modified with SB
We have performed and published first in human clinical trials using SB-modified T cells. These clinical trials utilized CD19-specific chimeric antigen receptor-expressing T cells gene modified using the SB system (DNA plasmids encoding for CD19RCAR transposon and SB11 transposase) to treat patients with CD19-positive B-cell malignancies [47]. The primary end points of the published study were to assess the safety, feasibility, and T-cell persistence of therapy using T cells genetically modified using the SB system. Among seven autologous and 19 allogeneic patients treated with CD19-specific SB gene modified T cells, excellent progression-free and overall survival were observed. There were no acute or late toxicities observed, nor was there exacerbation of graft-versus-host disease in the allogeneic patients. As expected 99.9% of SB-mediated CAR transgene integrated into TA dinucleotide sites, distributed evenly over each chromosome. This clinical trial also confirmed that the SB11 construct was not integrated into the host genome, and no gene hopping was observed. The lack of SB11 genes in the CAR T cell product was a release criteria before infusing the cells into patients (4 weeks after electroporation). No significant genotoxic events were observed. In summary, this study showed that the infusion of T cells genetically modified to express CAR by the SB system is feasible and safe in patients with B-cell malignancies.
Translational insight
Our initial clinical trials demonstrated the safety and feasibility of using the SB system for T cell gene therapy. However, to evaluate genotoxicity, especially validating that cells have no random integration of transposase from naked DNA plasmid, we had to culture genetically modified T cells for a relatively longer period (4 weeks). Although this protocol generated a large number of CAR-expressing T cells, the multiple in vitro stimulations caused differentiation of the T cells to several memory phenotypes (including effector memory, stem cell memory and central memory) and terminally differentiated effector cells. As the therapeutic effect correlates with the T cell differentiation stage (less differentiated T cells induce better therapeutic efficacy) in adoptive T cell therapy [48], it would be ideal to minimize the in vitro culture period. The shortening of culture period of SB mediated CAR T cells may be possible by use of hyperactive transposases (SB100x) from in vitro transcribed mRNA [35], which are not integrated into host genome, or using small therapeutic constructs (e.g. pFAR4 [45] or minicircle vector [39,46]). As SB-mediated CAR transduction can be done without prior T cell stimulation, the shortest manufacturing period of CAR T cells using the SB system can be shortened to within a day (‘minimally manipulated CAR T cells’ [Figure 3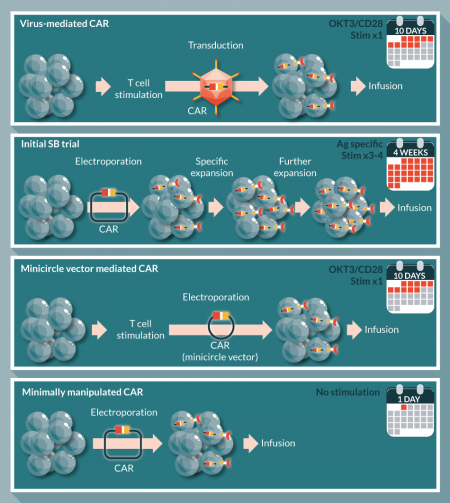
| Table 1: Versatility and cost–effectiveness of non-viral Sleeping Beauty-mediated gene transfer approach | |
|---|---|
| Non-viral Sleeping Beauty | Viral delivery |
| Target solid tumor intracellular neo-antigens via multiple TCRs | Limited appeal for targeting multiple intracellular antigens via TCRs |
| Cost-effective approach | High cost approach |
| Rapid manufacture | Labor intensive, slow manufacture |
| Customizable, able to swap in different receptors | Challenging to customize |
TCR gene therapy is another way to redirect T cell specificity to tumors. Although many tumor-associated antigens have been identified and targeted by TCR gene therapy, NYESO1/A2 may be the only promising target comparable with CAR T cell therapy [49,50]. Accumulated evidence suggests that tumor-specific, mutation-derived ‘neoantigens’ are promising targets [51] and are the predominant antigens recognized by T cells after immune-checkpoint modulation in highly mutation-loaded tumors [52]. As already exhausted neoantigen-specific T cells in the patient’s body are usually re-exhausted after invigoration by immune-checkpoint modulation and failed to become memory T cell, supplying less differentiated and non-exhausted neoantigen specific T cell would be desired [53]. Indeed TCR gene therapy against neoantigens would be a promising strategy to enhance anti-tumor immunity against highly mutation-loaded tumors [54]. Unfortunately, the majority of neoantigens identified so far are specific to the patient’s individual tumor cells, rather than being disease-related shared mutations (KRAS, p53, etc.). From this observation, TCR gene therapy against neoantigens will be likely patient-specific individualized therapy rather than disease-specific therapy like CAR T cells. If this is the case, rapid and timely production of therapeutic genes and TCR gene transferred T cells will be desired. The DNA plasmid-based SB system would be an optimal tool for using TCR gene transfer targeting a patient’s tumor-specific mutation-derived ‘neo-antigen’. We have established a platform to generate neoantigen-specific T cells by TCR gene transfer in a short time period. This platform consists of single-cell TCR sequencing (CDR3), a single reaction antigen-specific TCR construction in the SB plasmid system, and a high-throughput electroporation using Nucleofector®. We have also established a high-throughput TCR evaluation system using TCR-deficient reporter cells and a high-throughput flow cytometer. These systems have been tested with model antigens, and we will further test and refine this system by using patient-derived samples.
Financial and competing interests disclosure
Some of the technology described in this article was advanced through research conducted at the University of Texas MD Anderson Cancer Center (MD Anderson) by Laurence Cooper. In January 2015, the technology was licensed by MD Anderson for commercial application to ZIOPHARM Oncology, Inc., and Intrexon Corporation, in exchange for equity interests in each of these companies. The authors received equity as a result of the licensing of this technology. As of April 2016 Drs. Torikai and Moyes no longer have any equity interest. On May 7, 2015, Dr. Cooper was appointed as the Chief Executive Officer at ZIOPHARM. Dr Cooper is also a Visiting Scientist at MD Anderson. The information being reported in this publication is research in which The University of Texas MD Anderson Cancer Center has an institutional financial conflict of interest. Because MD Anderson is committed to the protection of human subjects and the effective management of its financial conflicts of interest in relation to its research activities, MD Anderson has implemented an Institutional Conflict of Interest Management and Monitoring Plan to manage and monitor the conflict of interest with respect to MD Anderson’s conduct of this research.
References
1. Blaese RM, Culver KW, Miller AD et al. T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science 1995; 270(5235): 475–80. CrossRef
2. Hacein-Bey-Abina S. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008; 118: 3132–42. CrossRef
3. Kohn DB, Sadelain M, Glorioso JC. Occurrence of leukaemia following gene therapy of X-linked SCID. Nat. Rev. Cancer 2003; 3(7): 477–88. CrossRef
4. Kalos M, Levine BL, Porter DL et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011; 3(95): 95ra73. CrossRef
5. Kebriaei P, Singh H, Huls MH et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J. Clin. Invest. 2016; 126(9): 3363–76. CrossRef
6. Johnson LA, June CH. Driving gene-engineered T cell immunotherapy of cancer. Cell Res. 2017; 27(1): 38–58. CrossRef
7. Jensen MC, Clarke P, Tan G et al. Human T lymphocyte genetic modification with naked DNA. Mol. Ther. 2000; 1(1): 49–55. CrossRef
8. Kay MA, Glorioso JC, Naldini L. Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat. Med. 2001; 7(1): 33–40. CrossRef
9. Mitchell RS, Beitzel BF, Schroder AR et al. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004; 2(8): E234. CrossRef
10. Howe SJ, Mansour MR, Schwarzwaelder K et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest. 2008; 118(9): 3143–50. CrossRef
11. Matrai J, Chuah MK, VandenDriessche T. Recent advances in lentiviral vector development and applications. Mol. Ther. 2010; 18(3): 477–90. CrossRef
12. Sinn PL, Sauter SL, McCray PB, Jr. Gene therapy progress and prospects: development of improved lentiviral and retroviral vectors–design, biosafety, and production. Gene Ther. 2005; 12(14): 1089–98. CrossRef
13. Naldini L. Gene therapy returns to centre stage. Nature 2015; 526(7573): 351–60. CrossRef
14. Newrzela S, Cornils K, Li Z et al. Resistance of mature T cells to oncogene transformation. Blood 2008; 112(6): 2278–86. CrossRef
15. Grupp SA, Kalos M, Barrett D et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013; 368(16): 1509–18. CrossRef
16. Porter DL, Hwang W-T, Frey NV et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015; 7(303): 303ra139-303ra139. DOI
17. Garfall AL, Maus MV, Hwang WT et al. Chimeric Antigen Receptor T Cells against CD19 for Multiple Myeloma. N. Engl. J. Med. 2015; 373(11): 1040–7. CrossRef
18. Kochenderfer JN, Dudley ME, Kassim SH et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015; 33(6): 540–9. CrossRef
19. Kochenderfer JN, Somerville RP, Lu T et al. Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated With High Serum Interleukin-15 Levels. J. Clin. Oncol. 2017: JCO2016713024. CrossRef
20. Davila ML. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014; 6:224ra225.
CrossRef
21. Newick K, O’Brien S, Moon E, Albelda SM. CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med. 2016; 68: 139–52.
CrossRef
22. Ledford H. T-cell therapy extends cancer survival to years. Nature 2014; 516(7530): 156. CrossRef
23. Wang X, Riviere I. Manufacture of tumor- and virus-specific T lymphocytes for adoptive cell therapies. Cancer Gene Ther. 2015; 22(2): 85–94. CrossRef
24. Copeland NG, Jenkins NA. Harnessing transposons for cancer gene discovery. Nat. Rev. Cancer 2010; 10(10): 696–706. CrossRef
25. Hackett PB, Largaespada DA, Cooper LJ. A transposon and transposase system for human application. Mol. Ther. 2010; 18(4): 674–83. CrossRef
26. Kawakami K. Tol2: a versatile gene transfer vector in vertebrates. Genome Biol. 2007; 8 Suppl. 1: S7. CrossRef
27. Ding S, Wu X, Li G, Han M, Zhuang Y, Xu T. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell 2005; 122(3): 473–83. CrossRef
28. Ivics Z, Hackett PB, Plasterk RH, Izsvak Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 1997; 91(4): 501–10. CrossRef
29. Manuri PV, Wilson MH, Maiti SN et al. piggyBac transposon/transposase system to generate CD19-specific T cells for the treatment of B-lineage malignancies. Hum. Gene Ther. 2010; 21(4): 427–37. CrossRef
30. Singh H, Manuri PR, Olivares S et al. Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system. Cancer Res. 2008; 68(8): 2961–71. CrossRef
31. Tsukahara T, Iwase N, Kawakami K et al. The Tol2 transposon system mediates the genetic engineering of T-cells with CD19-specific chimeric antigen receptors for B-cell malignancies. Gene Ther. 2015; 22(2): 209–15. CrossRef
32. Huang X, Wilber AC, Bao L et al. Stable gene transfer and expression in human primary T cells by the Sleeping Beauty transposon system. Blood 2006; 107(2): 483–91. CrossRef
33. Grabundzija I, Irgang M, Mates L et al. Comparative analysis of transposable element vector systems in human cells. Mol. Ther. 2010; 18(6): 1200–209. CrossRef
34. Maiti SN, Huls H, Singh H et al. Sleeping beauty system to redirect T-cell specificity for human applications. J. Immunother. 2013; 36(2): 112–23. CrossRef
35. Jin Z, Maiti S, Huls H et al. The hyperactive Sleeping Beauty transposase SB100X improves the genetic modification of T cells to express a chimeric antigen receptor. Gene Ther. 2011; 18(9): 849–56. CrossRef
36. Mates L, Chuah MK, Belay E et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat. Genet. 2009; 41(6): 753–61. CrossRef
37. Xue X, Huang X, Nodland SE et al. Stable gene transfer and expression in cord blood-derived CD34+ hematopoietic stem and progenitor cells by a hyperactive Sleeping Beauty transposon system. Blood 2009; 114(7): 1319–30. CrossRef
38. Wilber A, Linehan JL, Tian X et al. Efficient and stable transgene expression in human embryonic stem cells using transposon-mediated gene transfer. Stem Cells 2007; 25(11): 2919–27. CrossRef
39. Monjezi R, Miskey C, Gogishvili T et al. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia 2017; 31(1): 186–94. CrossRef
40. Peng PD, Cohen CJ, Yang S et al. Efficient nonviral Sleeping Beauty transposon-based TCR gene transfer to peripheral blood lymphocytes confers antigen-specific antitumor reactivity. Gene Ther. 2009; 16(8): 1042–9. CrossRef
41. Gogol-Döring A, Ammar I, Gupta S et al. Genome-wide Profiling Reveals Remarkable Parallels Between Insertion Site Selection Properties of the MLV Retrovirus and the piggyBac Transposon in Primary Human CD4+ T Cells. Mol. Ther. 2016; 24(3): 592–606. CrossRef
42. Izsvak Z, Ivics Z, Plasterk RH. Sleeping Beauty, a wide host-range transposon vector for genetic transformation in vertebrates. J. Mol. Biol. 2000; 302(1): 93–102. CrossRef
43. de Silva S, Mastrangelo MA, Lotta LT, Jr., Burris CA, Federoff HJ, Bowers WJ. Extending the transposable payload limit of Sleeping Beauty (SB) using the Herpes Simplex Virus (HSV)/SB amplicon-vector platform. Gene Ther. 2010; 17(3): 424–31. CrossRef
44. Zayed H, Izsvak Z, Walisko O, Ivics Z. Development of hyperactive sleeping beauty transposon vectors by mutational analysis. Mol. Ther. 2004; 9(2): 292–304. CrossRef
45. Thumann G, Harmening N, Prat-Souteyrand C et al. Engineering of PEDF-Expressing Primary Pigment Epithelial Cells by the SB Transposon System Delivered by pFAR4 Plasmids. Mol. Ther. Nucleic Acids 2017; 6: 302–14. CrossRef
46. Sharma N, Cai Y, Bak RO, Jakobsen MR, Schroder LD, Mikkelsen JG. Efficient sleeping beauty DNA transposition from DNA minicircles. Mol. Ther. Nucleic Acids 2013; 2: e74. CrossRef
47. Kebriaei P, Huls H, Jena B et al. Infusing CD19-directed T cells to augment disease control in patients undergoing autologous hematopoietic stem-cell transplantation for advanced B-lymphoid malignancies. Hum. Gene Ther. 2012; 23(5): 444–50. CrossRef
48. Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat. Rev. Cancer 2012; 12(10): 671–84. CrossRef
49. Robbins PF, Kassim SH, Tran TL et al. A Pilot Trial Using Lymphocytes Genetically Engineered with an NY-ESO-1-Reactive T-cell Receptor: Long-term Follow-up and Correlates with Response. Clin. Cancer Res. 2014; 21(5): 1019–27.
CrossRef
50. Rapoport AP, Stadtmauer EA, Binder-Scholl GK et al. NY-ESO-1–specific TCR–engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015; 21(8): 914–21. CrossRef
51. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015; 348(6230): 69–74. CrossRef
52. McGranahan N, Furness AJS, Rosenthal R et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016; 46(4): 577–86. CrossRef
53. Pauken KE, Sammons MA, Odorizzi PM et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016; 354(6316): 1160–5. CrossRef
54. Leisegang M, Kammertoens T, Uckert W, Blankenstein T. Targeting human melanoma neoantigens by T cell receptor gene therapy. J. Clin. Invest. 2016; 126(3): 854–8. CrossRef
55. Hurton LV, Singh H, Switzer KC et al. Very Rapid Production of CAR+T-Cells upon Non-Viral Gene Transfer Using the Sleeping Beauty System. Blood 2016; 128(22): 2807. Website
Affiliations
Hiroki Torikai1, Judy S Moyes1 & Laurence JN Cooper1, 2*
1 The University of Texas, MD Anderson Cancer Center, Division of Pediatrics, TX, USA
2 Ziopharm Oncology, Inc.
*Author for correspondence: ljncooper@ziopharm.com

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.