The road to gene therapy for recessive dystrophic epidermolysis bullosa
Cell Gene Therapy Insights 2017; 3(2), 75-81.
10.18609/cgti.2017.011
The road towards development of an optimal gene therapy for recessive dystrophic epidermolysis bullosa has been long and many lessons have been learned along the way. This editorial highlights the key advances and challenges in the field as it progress towards clinical translation.



Severe generalized recessive dystrophic epidermolysis bullosa (RDEB) has long been recognized as one of the most debilitating subtypes of the family of blistering diseases known as epidermolysis bullosa (EB). It is characterized by painful blistering and scarring due to absence of the basement membrane protein type VII collagen (C7). Despite the critical need for corrective therapy, the current medical standard of care is purely supportive, centering on wound care, nutrition and treating infections when they arise. The recent publication ‘Safety and Wound Outcomes Following Genetically Corrected Autologous Epidermal Grafts in Patients With RDEB’ [1] is the first clinical trial to directly address RDEB at the molecular level, using ex vivo gene therapy to restore C7 expression and reverse the severe blistering skin disease in patients with RDEB.
The pathway towards a Phase 1 clinical trial of gene therapy for EB had been a long journey, built upon a large foundation of previous work. It’s taken over 140 years, since EB was first recognized [2] , to fully uncover the molecular etiology of the individual EB subtypes, with some rare variants of this disorder having been molecularly characterized only in the last few years [3]. The first breakthrough on the molecular etiology of dystrophic EB came with the discovery of C7 [4], which was shown to localize to anchoring fibrils [5], ultrastructural entities that wrap around interstitial collagen fibrils and appear to connect the papillary dermis to the basement membrane [6]. C7 was then discovered to be missing from the skin of patients with severe RDEB [7] implicating C7 defects in the pathogenesis of RDEB. Following the cloning of COL7A1, the human C7 gene [8], COL7A1 mutations were identified in RDEB patients [9,10], setting the stage for genetic molecular correction RDEB.
Gene replacement therapy for EB was first performed in a single case study of a junctional EB patient caused by missense mutation of the LAMB3 gene [11]. The LAMB3 gene, following retroviral gene transfer, continued to show protein expression in patient skin even after 6.5-year follow-up [12]. Following the treatment of this junctional EB (JEB) patient, other reports of retroviral-mediated insertional mutagenesis arose associated with the development of leukemia after gene therapy of severe combined immunodeficiency patients [13,14]. However, retrovirally engineered skin grafts had key advantages over virally treated immune cells in that suspicious skin lesions could be more easily monitored, biopsied and removed. This added measure of safety contributed towards approval of retroviral keratinocyte graft therapy by the US Recombinant DNA Advisory Committee in 2007.
Compared to retroviral JEB studies, retroviral therapy for RDEB proved more challenging due to the extremely large size of the COL7A1 gene and the difficulty of generating a high titer retroviral vector. This was addressed by the systemic removal of non-essential viral elements, to create enough space to accommodate the large COL7A1 gene, several mutations within the viral packaging signal and choice of the virus envelop protein. This ultimately proved successful in preclinical studies, demonstrating basement membrane associated C7 expression for up to 1 year in primary regenerated human RDEB skin equivalents xenografted to immunodeficient mice [15].
Having achieved pre-clinical objectives for RDEB gene therapy, the next step on the road to clinical trials was to make the transition from laboratory grade to a clinical grade manufacturing process. First, it was necessary to produce recombination free master cell bank and Good Manufacturing Process (GMP) grade retrovirus with similar titer to the virus used in the laboratory. At the same time, animal-derived culture reagents and supplements deemed to be free of adventitious viruses, including bovine spongiform encephalopathy and others, had to be sought for and tested to support robust graft production.
Figure 1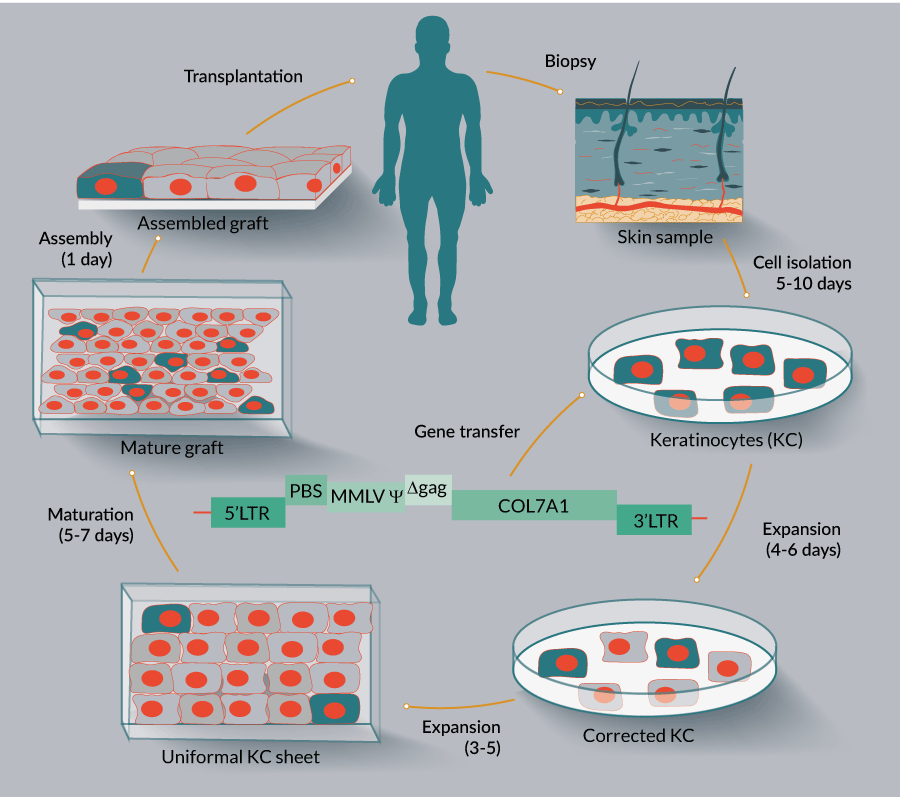
For these studies, a special graft production facility needed to be established and maintained, which, although was not certifiably GMP,nevertheless adhered to many GMP requirements, such as a low air particle count supported by a HEPA filtration unit, material quality control, equipment monitoring, etc. Numerous protocols and test readouts needed to be sequentially followed during the graft manufacturing process as required by the US FDA, and as overseen by a data safety monitoring board, including formation of the recombinant competent retrovirus, safety, sterility, endotoxin and mycoplasma levels. The study demonstrated durable expression of C7 in the grafts, following placement on the patient in the OR, resulting in linear incorporation of the therapeutic gene product into the basement membrane, specifically into anchoring fibrils as shown by immune-electron microscopy. An important part of this study was demonstrating expression of C7 using both NC1 and NC2 domain antibodies. One important advance was in demonstrating that the monoclonal antibody LH24 [16]recognized a portion of the C7 molecule near the NC2 domain[1].
Previous studies of C7 molecular delivery all relied solely on reactivity with C7 NC1 domain antibodies and could not rule out the possibility that what was purported to be full length C7 could have been degraded NC1 domain fragments. The Siprashvili et al. study confirmed, in fact, that stable NC1 containing degradation products of C7 do indeed exist [1]. Therefore, the LH24 antibody will prove useful in future molecular therapies to demonstrate, in combination with NC1 antibodies, the presence of intact C7 in tissues.
Immune reaction to a therapeutic gene product is an important concern, particularly in null patients. Patients with EB acquisitia (EBA), a group of patients with acquired C7 autoimmunity, demonstrate a predominance of autoantibodies against what appeared to be the most antigenic domain on the C7 molecule, the NC1 domain [17]. For this reason, it was hypothesized that RDEB patients who expressed even small amounts of NC1 domain might have a reduced chance of developing an autoimmune reaction to the full length C7 therapeutic gene product compared to RDEB patients who showed no expression at all.
Due to the finding of negative reactivity with various C7 antibodies by indirect immunofluorescence microscopy, most RDEB patients were traditionally considered as C7 null. However, in preparation for gene therapy clinical trials, expression of C7 was examined in cultured RDEB patients’ skin cells using a more sensitive Western blot assay. While it was previously established that both fibroblasts and keratinocytes produce C7 and deposit it into the basement membrane [18], fibroblasts downregulate C7 to low levels after in vitro passaging [18], making RDEB keratinocytes more optimal for this assay. From these studies it was established that the majority of RDEB patients, even though they displayed negative staining for C7 on IDIF, are technically not true nulls because their keratinocytes demonstrated low but detectable expression of a fragment of C7 containing the large noncollagenous 1 (NC1) domain [19]. Thus it was reasoned that NC1+ patients’ immune systems would have a better chance of recognizing the antigenic C7 NC1 as self, reducing the chance of immunologic reactions following gene therapy. Thus, the lack of baseline C7 antibodies and the demonstration of positive expression of C7 NC1 domain by Western blot of patient keratinocyte cultures became two key inclusion criteria for the study.
Following placement on chronic wounds, durable C7 expression was demonstrated in genetically engineered grafts, showing expression even at the 1-year point, the longest time measured in the study. Coincident with the molecular correction was the observation of clinical improvement and resistance to wounding and blistering noted both by global investigator assessments, photographic software analysis, as well as in the patients’ reports of the skin feeling stronger. One patient was able to walk much further following placement of grafts on his feet, while another found that he could finally sleep on his side, following placement of a graft over a chronic erosion on his shoulder. However, there was variability from patient to patient and both C7 expression and clinical efficacy gradually decreased over time.
Several factors may have contributed to the gradual decline of C7 expression and variability from patient to patient. One factor that appeared to play a role in the trial was immobilization of the graft following placement in the OR. Grafts placed on limbs could be more easily immobilized and protected from pressure or mechanical disruption, and generally proved more successful than grafts placed over areas more subject to trauma such as the back. An additional theoretical factor affecting graft durability was the possibility of non-transduced stem cells in the wound bed growing and then displacing/undermining the transduced cells in the graft. It is possible that controlled depth laser destruction of the wound bed, prior to grafting, such as that performed by the JEB retroviral study [11],could help to address this issue.One other possible factor is limitation of available stem cells during graft production. The previous study of JEB patients utilized the palm as a rich source of skin stem cells; however, the scarring pseudosyndactyly compromised available stem cells in RDEB patients palms. Instead stem cell rich areas of skin such as the scalp could be considered for graft production.
Immune surveillance was of critical importance in this trial. This is illustrated well in the findings noted in patient four. Immunofluorescence (IF) microscopy revealed patient four’s serum immune reactivity to C7 to be negative prior to grafting, but positive after grafting. To further investigate, patient pre-grafting sera was further tested using a more sensitive Western blot assay, revealing that the patient indeed demonstrated pre-graft C7 antibodies. The finding of pre-graft antibodies suggested, in this case, that C7 gene therapy exacerbated a preexisting immune response rather than causing a de novo immune reaction. As a result of the findings with patient four, the protocol was subsequently changed to include Western blot evaluation of all patient sera pre- and post-grafting.
These observations bring to light an important point. Previous studies have documented the occurrence of anti-C7 antibodies in RDEB patients[20]; however, because these antibodies did not react with the skin by IF microscopy, it was suggested that these antibodies may be inconsequential and non-pathogenic. The results of the current gene therapy clinical trial, however, suggest that these low levels of circulating C7 antibodies may be predictive of a potential exacerbating immune response following exposure to therapeutic C7. Thus C7 antibodies in RDEB patients may have more physiological relevance than was previously thought, especially in regards to prospective C7 molecular therapies.
In summary, the road towards development of an optimal gene therapy for EB has been long and many lessons have been learned along the way. In addition, many lessons still need to be learned. The current next steps towards translational development of C7 gene therapy as applied to engineered keratinocyte grafts will involve demonstration of effectiveness and safety of this treatment in larger numbers of patients, particularly in the pediatric population, towards the goal of FDA approval of this molecular correction therapy to improve the pain, debilitation and quality of life for patients afflicted by RDEB.
FINANCIAL & COMPETING INTERESTS DISCLOSURE
The authors have no relevant financial involvement with an organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock options or ownership, expert testimony, grants or patents received or pending, or royalties. No writing assistance was utilized in the production of this manuscript.
References
1. Siprashvili Z, Nguyen NT, Gorell ES et al. Safety and Wound Outcomes Following Genetically Corrected Autologous Epidermal Grafts in Patients With Recessive Dystrophic Epidermolysis Bullosa. JAMA 2016; 1808–17.
2. Hebra Fv. Arztlicher Bericht des K.K allegemeinen Krankenhauses zu Wien vom Jare 1870. Vienna 1870.
3. Fine JD, Bruckner-Tuderman L, Eady RA et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J. Am. Acad. Dermatol. 2014; 1103–26.
4. Burgeson RE, Morris NP, Murray LW et al. The structure of type VII collagen. Ann. NY Acad. Sci. 1985; 47–57.
5. Sakai LY, Keene DR, Morris NP, Burgeson RE. Type VII collagen is a major structural component of anchoring fibrils. J. Cell Biol. 1986; 1577–86.
6. Keene DR, Sakai LY, Lunstrum GP, Morris NP, Burgeson RE. Type VII collagen forms an extended network of anchoring fibrils. J. Cell Biol. 1987; 611–21.
7. Bruckner-Tuderman L, Mitsuhashi Y, Schnyder UW, Bruckner P. Anchoring fibrils and type VII collagen are absent from skin in severe recessive dystrophic epidermolysis bullosa. J. Invest. Dermatol. 1989; 3–9.
8. Parente MG, Chung LC, Ryynanen J, Uitto J. Human type VII collagen: cDNA cloning and chromosomal mapping of the gene (COL7A1) on chromosome 3 to dominant dystrophic epidermolysis bullosa. Am. J. Hum. Genet. 1991; 119–35.
9. Hilal L, Rochat A, Duquesnoy P et al. A homozygous insertion-deletion in the type VII collagen gene (COL7A1) in Hallopeau-Siemens dystrophic epidermolysis bullosa. Nat. Genet. 1993; 287–93.
10. Christiano AM, Greenspan DS, Hoffman GG et al. A missense mutation in type VII collagen in two affected siblings with recessive dystrophic epidermolysis bullosa. Nat. Genet. 1993; 62–6.
11. Mavilio F, Pellegrini G, Ferrari S et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat. Med. 2006; 1397–402.
12. De Rosa L, Carulli S, Cocchiarella F et al. Long-term stability and safety of transgenic cultured epidermal stem cells in gene therapy of junctional epidermolysis bullosa. Stem Cell Reports 2014; 1–8.
13. Howe SJ, Mansour MR, Schwarzwaelder K et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest. 2008; 3143–50.
14. Hacein-Bey-Abina S, Garrigue A, Wang GP et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008; 3132–42.
15. Siprashvili Z, Nguyen NT, Bezchinsky MY et al. Long-term type VII collagen restoration to human epidermolysis bullosa skin tissue. Hum. Gene Ther. 2010; 1299–310.
16. Sinclair R, Wojnarowska F, Leigh I, Dawber R. The basement membrane zone of the nail. Br. J. Dermatol. 1994; 499–505.
17. Lapiere JC, Woodley DT, Parente MG et al. Epitope mapping of type VII collagen. Identification of discrete peptide sequences recognized by sera from patients with acquired epidermolysis bullosa. J. Clin. Invest. 1993; 1831–9.
18. Marinkovich MP, Keene DR, Rimberg CS, Burgeson RE. Cellular origin of the dermal-epidermal basement membrane. Dev. Dyn. 1993; 255–67
19. Gorell ES, Nguyen N, Siprashvili Z, Marinkovich MP, Lane AT. Characterization of patients with dystrophic epidermolysis bullosa for collagen VII therapy. Br. J. Dermatol. 2015; 821–3.
20. Woodley DT, Cogan J, Wang X et al. De novo anti-type VII collagen antibodies in patients with recessive dystrophic epidermolysis bullosa. J. Invest. Dermatol. 2014; 1138–40.
AFFILIATIONS
M Peter Marinkovich1,2, Ngon T Nguyen1 & Zurab Siprashvili1
1Department of Dermatology, Stanford University School of Medicine, Stanford, CA 94305, USA
2Dermatology Service, Veteran’s Affairs Medical Center, Palo Alto, CA 94304, USA

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.