Specifically integrating vectors for targeted gene delivery: progress and prospects
Cell Gene Therapy Insights 2017; 3(2), 103-123.
10.18609/cgti.2017.013
Integrating vectors based on viruses or transposons are efficient gene delivery vehicles and promising tools for gene therapy. While different vector systems have different preferences and biases when it comes to target site selection, integration can always occur at vast numbers of potential sites throughout the human genome. This can result in unpredictable expression of the transgene (position effects), and can disrupt host genes or regulatory elements (genotoxicity), thereby potentially causing malignant transformations. Our knowledge about the natural target site selection properties of these gene insertion systems can be translated into artificial, experimental retargeting with the goal of introducing a bias into their insertion profiles. Here, we provide an overview of naturally occurring targeting mechanisms of viruses and transposons, and of the different molecular strategies that have been followed to manipulate their target site selection to derive stably integrating vectors with enhanced safety profiles.
Integrating vector systems
The advent of gene therapy has significantly transformed the way we treat human genetic defects. For most of human history, treating genetic diseases meant attempting to ameliorate the symptoms. However, the ability to modify the human genome has made it possible to correct the underlying genetic defects. Apart from replacing defective versions of genes or regulatory elements, the same techniques can also be used to introduce novel functions to cells, for example for use in cellular therapies. Stable introduction of genes, whether to replace a defective gene or to introduce a new function, is dependent on technologies that can integrate these pieces of DNA into the genome. Without genomic integration, the therapeutic gene is only transiently expressed and a treatment would have to be repeated on a regular basis. So-called integrating vectors are often based on viruses, which possess the natural ability to transfer DNA across the cell membrane and integrate it into the host genome. Non-viral integrating systems, for example transposon-based vectors, are also able to stably integrate DNA into target genomes, but need to be first introduced into the cell.
After initial enthusiasm, it became clear that gene therapy using integrating vectors is associated with considerable risks. Probably the most prominent cases were the studies attempting to treat X-linked severe combined immunodeficiency (X-SCID) using autologous hematopoietic stem cells, which were modified ex vivo using first-generation γ-retroviral vectors based on the murine leukemia virus (MLV) [1]. While the treatment successfully corrected the defect in almost all patients, five out of 20 developed leukemia within approximately 5 years [2–4]. These adverse events were shown to be related to insertion of the transgene near the LMO2 gene and subsequent overexpression of LMO2 induced by the retroviral long terminal repeats (LTRs) [5], although other, insertion-unrelated events were also involved [6]. The development of leukemia after γ-retroviral gene therapy was also reported from trials treating other diseases [7,8].
Despite insertion-related complications, gene therapy was more efficient at treating X-SCID than hematopoietic stem cell transfer [9] and the survival rates of both therapies are similar [10]. Newer γ-retroviral vectors have been modified to be less genotoxic [11]. Clinical trials using integrating vectors have shown promise for the treatment of several diseases, including adrenoleukodystrophies [12,13], human immunodeficiency virus (HIV) infection [14], β-thalassemia [15], Wiskott–Aldrich syndrome [16,17] and B-cell malignancies [18–21]. In 2012, the first gene therapy product, marketed under the name Glybera, was approved in Europe; Glybera is an adeno-associated virus (AAV)-based vector for treatment of lipoprotein lipase deficiency. The first retrovirus-based treatment based on stable gene transfer into hematopoietic stem cells – called Strimvelis – was approved in 2016 to treat severe combined immunodeficiency due to adenosine deaminase deficiency (ADA-SCID).
The development of leukemias highlights an intrinsic problem of many integrating vectors: their integration can occur at large numbers of sites scattered around the genome (which is already potentially mutagenic) with vector-specific integration biases towards actively transcribed genes and their regulatory elements (which makes some vector systems even more mutagenic). For example, lentiviral vectors and γ-retroviral vectors actively target transcription units or transcriptional regulatory elements of genes, respectively [22–24]. Some vectors based on transposons, such as the piggyBac (PB) element, have integration profiles similar to viruses[25]. The Sleeping Beauty (SB) system, on the other hand, has been found to integrate in a close-to-random manner with only a small bias towards genes [25–29]. Indeed, when compared directly to MLV-based γ-retroviral vectors, HIV-based lentiviral vectors and the PB transposon in human CD4+ T cells, the SB transposon was found to display the least deviation from random with respect to genome-wide distribution: no apparent bias was seen for either heterochromatin marks or euchromatin marks and only a weak correlation with transcriptional status of targeted genes was detected [25]. However, even vectors with a completely random integration profile can insert into or near genes by chance.
Random integration of transgenes into target genomes can have two consequences, both of which are highly problematic for gene therapy applications (Figure 1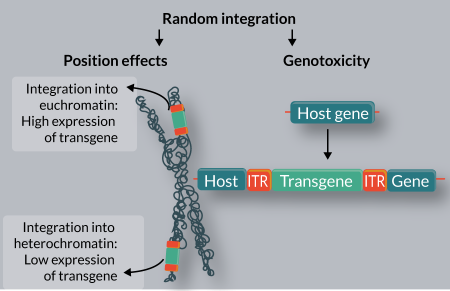
Naturally occurring targeted insertion
When attempting to retarget vectors, it is possible to mimic nature, as some naturally occurring viruses and transposons have site- or region-specific insertion preferences (Table 1).
| Table 1: Naturally occurring targeted insertion systems. | ||||
|---|---|---|---|---|
| Recombinase | Origin | Target | Cofactors | Ref. |
| Lentiviral IN | Lentiviruses | Transcription units | LEDGF | [32–35] |
| γ-retroviral IN | γ-retroviruses | TSSs, CpG islands | BET proteins | [44–46] |
| AAV Rep | AAV | AAVS1 site | None | [50–52] |
| Ty1 IN | S. cerevisiae | Upstream of Pol III transcribed genes | TFIIIB of Pol III | [61–65] |
| Ty3 IN | S. cerevisiae | Upstream of Pol III transcribed genes | TFIIIB, TFIIIC of Pol III | [66–68] |
| Ty5 IN | S. cerevisiae | Heterochromatin | Sir4p | [76–81] |
| Tf1 IN | S. pombe | Promoters of Pol II transcribed genes, arrested replication fork | Sap1 | [72,73,75] |
| TRE ORF1 | Dictyostelium | tRNA genes | Pol III | [69–71] |
| Tn7 transposase | Bacterial | Replicating DNA, attTn7 | β clamp, TnsD, TnsE | [84,86–88] |
| PB transposase | Insects | TSSs | BET proteins | [26] |
| SB transposase | Fish | DNA sequences that resemble the transposase binding sites | The SB transposase itself | [26] |
| ΦC31 IN | ΦC31 phage | Pseudo attP sites in the human genome | None | [92] |
Lentiviruses like HIV preferentially integrate into active transcription units [22]. This targeting effect is based on interaction between the viral integrase (IN) and the host chromatin reader lens epithelium-derived growth factor (LEDGF) (Figure 2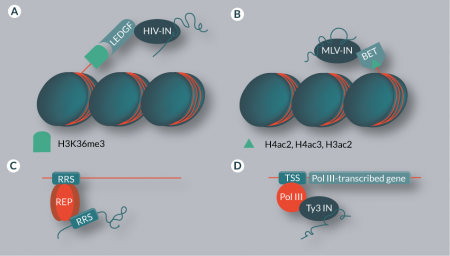
Insertions of MLV and γ-retroviral vectors are targeted towards transcription start sites (TSSs), CpG islands and DNAse I-hypersensitive sites [23,42,43]. This characteristic bias has been shown to be the result of an interaction between host factors of the bromodomain and extraterminal domain (BET) family and the viral IN (Figure 2B) [44–46]. BET proteins are chromatin readers and recognize di- and triacetylated H4 and diacetylated H3 [47]. Disruption of the BET interaction domain of the MLV IN randomizes MLV integration distribution, thereby improving the safety profile of γ-retroviral vectors by ‘de-targeting’ [48]. Although this approach cannot be used to derive an integration profile that would be safer than completely random, it has the appeal that no overexpression of targeting proteins (see below) is required to achieve a change in chromosomal integration patterns [47].
While lentiviruses and γ-retroviruses are targeted to elements that occur in the human genome many times, integration of wild-type AAV is site-specific. AAV integrates into the AAVS1 locus located on human chromosome 19 [49]. The viral Rep protein simultaneously binds Rep recognition sequences (RRSs) in the viral inverted terminal repeats (ITRs) as well as in the human genome, bringing the integration complex in close vicinity to the target site (Figure 2C) [50–52]. Such target site specificity, combined with a lack of pathogenicity of AAV, would be highly advantageous for gene therapy applications [53]. However, in recombinant AAV vectors the Rep gene is replaced by a genetic cargo, and thus the resulting vectors, unfortunately, lack targeted genomic integration [54]. Additionally, AAV proteins have been shown to cause immune complications [55].
The preference of several viruses to insert near actively transcribed genes might be related to the fact that viral genomes need to be transcribed after integration for their propagation. Thus, viruses that preferentially integrate in genomic regions accessible to the transcription machinery likely gain an evolutionary advantage [24]. Transposons, on the other hand, lack an extracellular phase in their life cycle [56]. This means that integrations that disrupt the host cell’s function are deleterious for the survival of the transposon itself. Consequently, many transposons have a lower preference for genes and transcription units than viruses [57].
The Saccharomyces cerevisiae retrotransposons Ty1 and Ty3 preferentially integrate into genomic regions upstream of RNA polymerase III (Pol III) TSSs [58,59]. While this might seem to contradict the general rule that transposons have a lower preference for transcriptionally active loci, it should be noted that the regions upstream of Pol III-transcribed genes are often gene-poor [60]. Ty1 has a particularly strong preference for tRNA genes and the 5S RNA gene, usually integrating in a window that extends several hundred base pairs upstream of the TSS [61–63]. An interaction between the Ty1 IN and the TFIIIB component of Pol III is responsible for this effect [64,65]. Ty3 also targets Pol III start sites, but integrations are found in a narrow window of one or two base pairs upstream of the TSS [57], and the TFIIIB and TFIIIC components of Pol III complexes are involved in the recruitment of Ty3 (Figure 2D) [66–68]. Other retrotransposons that specifically integrate near tRNA genes include the TRE (tRNA gene-targeting retrotransposable elements) elements from Dictyostelium discoideum, which are targeted via interaction with Pol III transcription factors [69–71]. The Tf1 retrotransposon in Schizosaccharomyces pombe preferentially integrates into promoters of Pol II-transcribed genes [72,73], and a major determinant of this target site selection is the DNA-binding protein Sap1, which binds to clusters of a 5-bp sequence motif [74]. Interaction of Sap1 with the Tf1 IN has been suggested to play an important role in tethering the preintegration complex – which consists of transposon DNA, IN and cofactors – to target sites [75]. In addition, it was recently shown that Sap1 guides Tf1 insertions into arrested replication forks [75]. In contrast to Ty1 and Ty3, the Ty5 yeast retrotransposon mostly integrates into heterochromatin [76–78]. This insertion preference is mediated via an interaction between a C-terminal domain of Ty5 IN and the host factor Sir4p [79–81]. Sir4p is a chromatin component predominantly found in telomeric heterochromatin [82,83]; thus, the interaction between Sir4p and the IN increases the likelihood of insertions into these regions.
Not only retrotransposons, but also DNA transposons can be targeted to certain sites and genomic regions in their hosts. For example, the Tn7 bacterial DNA transposon is capable of both DNA sequence- and structure-specific targeting, i.e., targeting specific nucleotide sequences or DNA structures independently of their nucleotide sequence. Tn7 transposition is targeted into actively replicating DNA by a mechanism involving the transposon-encoded protein TnsE [84], which interacts with the β clamp processivity factor of the DNA replication machinery [85]. Alternatively, another transposon-encoded factor, TnsD, binds to specific nucleotide sequences called attTn7 sites [86,87]. Targeting of Tn7 depends on which cofactor is used during integration [88]. Apart from the genome of the natural bacterial host, a small number of attTn7-like sites can be found in the human genome as well, but Tn7 transposition into these sites has not been established [89]. Finally, the eukaryotic DNA transposon PB, originally isolated from the cabbage looper moth, was recently shown to be targeted to TSSs through an interaction of the PB transposase with BET domain proteins, similar to the mechanism shown to be responsible for the enrichment of MLV integrations into TSSs [25].
A unifying theme in the targeting mechanisms described above is that a DNA- or chromatin-associated factor recruits preintegration complexes to certain genomic sites by physically interacting with a virus- or transposon-encoded protein. Another mechanism of targeted gene insertion exists that is based on direct recognition and interaction of the recombinase with a given DNA sequence in the genome. For example, the ΦC31 IN from a Streptomyces phage [90] mediates unidirectional recombination between the attP site of the phage genome and the attB site in the bacterial host genome, but it is also active in human cells [91]. Since the recognition sequences are relatively short (<40 bp), a number of sites with high similarity are expected to occur in the human genome. Indeed, a number of pseudo attP sites were found, and it was shown that ΦC31 IN can integrate DNA into these sites in a directed manner [92]. This makes the ΦC31 system an interesting tool for gene therapy, and it has been tested in preclinical models both ex vivo and in vivo [93–98]. However, it has been shown that expression of the ΦC31 IN can result in a DNA damage response and chromosomal aberrations, limiting its utility for therapeutic applications [99,100].
Targeting based on pseudo sequences in the human genome has also been observed for the SB transposon system, originally isolated from fish genomes [101]. Namely, it has been shown that SB integrations are enriched near genomic sequences that resemble the transposase binding sites that are normally found in the ITRs of the SB transposon [25]. Because SB transposase molecules likely interact with one another during the transposition process, binding of transposase molecules to these pseudo SB sites might tether transpositionally active transposase molecules bound to the transposon ITRs to these sites, thereby resulting in a fraction of insertions in their vicinity [25]. This naturally occurring tethering mechanism resembles targeted integration of wild-type AAV into the AAVS1 locus (simultaneous binding of Rep to the viral ITRs and to the genomic target site (Figure 2C)), and provides the molecular basis of artificial retargeting with the N-terminal N57 domain of the SB transposase (described below).
Artificial retargeting
In order to avoid position effects and insertional mutagenesis, it is of great interest to establish technologies that artificially retarget otherwise semi-randomly integrating vector systems to a precisely defined genomic region or specific sequence. Artificially retargeted vectors are not only useful for gene therapy. For example, fusions with DNA-binding domains (DBDs) of unknown specificity can be used to determine the binding sites of these domains by analyzing the integration profile of the retargeted vector [102–104].
Instead of relying on chance occurrence of pseudo sites in the human genome that are recognized by a recombinase enzyme (described above for the ΦC31 IN), custom DBDs that can be engineered to specifically interact with practically any sequence in the human genome are of great utility. In previous decades, two major classes of engineered DNA-binding proteins have been used for site-specific genome engineering: zinc finger proteins (ZFPs) [105] and transcription activator-like effectors (TALEs) [106]. In their most widespread applications, both ZFPs and TALEs serve as DBDs directly fused to an effector endonuclease domain derived from the restriction enzyme FokI. A revolutionary, programmable new tool is the CRISPR/Cas nuclease system [107], which relies on an RNA molecule and DNA:RNA base pairing for providing specificity for the cleavage reaction.
An overview of experimental approaches to retargeting is provided in Table 2 (targeting via direct recombinase fusions) and Table 3 (targeting via adapter proteins). Concerning the experimental systems, in vitro refers to cell-free assays. For cell culture assays, it is indicated whether integration was analyzed on a target plasmid or in the genome.
| Table 2: Retargeting via direct recombinase-DBD fusions. | ||||
|---|---|---|---|---|
| Hybrid protein | Targeting effect | Activity | System | Ref. |
| Viral vectors | ||||
| HIV-IN/LexA | Enrichment near LexA binding site | Like wild-type | In vitro | [115] |
| HIV-IN/lR | Enrichment near lR binding site | Like wild-type | In vitro | [116] |
| HIV-IN/Zif268 | Hotspots near Zif268 binding site | Integration like wild-type, abolished infectivity | In vitro | [117] |
| HIV-IN/E2C | 32% (six-fold increase) within 30bp of target | Like wild-type | In vitro | [118] |
| HIV-IN/E2C | 1.45% (ten-fold increase) near erbB-2 | <24% of wild-type | Cell culture (genomic) | [119] |
| ASV-IN/LexA | Hotspots <23 bp from LexA binding site | Similar to wild-type | In vitro | [120] |
| MLV-IN/Sp1 | Ca. 13% near Sp1 binding sites | Like wild-type | Cell culture (genomic) | [121] |
| Transposon vectors | ||||
| SB/Gal4 | 25% (11-fold increase) in 443 bp window | 26% of wild-type activity | Cell culture (plasmid) | [111] |
| SB/E2C | 17.8% (8-fold increase) in 443 bp window | 20% of wild-type activity | Cell culture (plasmid) | [111] |
| SB/E2C | Up to 2% of clones with integration near endogenous target site | <5% of wild-type activity | Cell culture (genomic) | [113] |
| SB/ZF-B | Up to 44.8% insertions into L1 elements | <10% of wild-type activity | Cell culture (genomic) | [113] |
| SB/Rep | Up to 2-fold enrichment 5kb from consensus Rep-binding sequence | 20–80% of wild-type activity | Cell culture (genomic) | [25] |
| PB/CHK2-ZFP | 50% enrichment in 500bp window around target site | Like wild-type | Cell culture (plasmid) | [126] |
| PB/Gal4 | 4.5-fold increase (24%) within 800bp of endogenous target sites | Like wild-type | Cell culture (genomic) | [110] |
| PB/TALE | <1% within 250kb of endogenous target site | Like wild-type | Cell culture (genomic) | [109] |
| IS30/cI | Tenfold increase in target plasmid | Similar to wild-type | E. coli (plasmid) | [127] |
| IS30/Gli1 | Several insertions near target site, but mostly illegitimate | Significantly reduced | Zebrafish (plasmid) | [127] |
| Mos1/Gal4 | 96% within 1kb of binding site | Transposition increased >ten-fold | Mosquito embryos (plasmid) | [128] |
| ISY100/Zif268 | Hotspot 7-17bp from binding site | Up to nine-fold lower than wild-type | E. coli (plasmid) | [129] |
| Other recombinases | ||||
| Tn3 resolvase/ Zif268 | Up to 100% recombination with appropriate target site | Like wild-type | E. coli (plasmid) | [131] |
| Gin recombinase/ dCas9 | Up to 32% recombination on plasmid target, <1% for genomic deletion | Depends on target sequence | Cell culture (plasmid, genomic) | [135] |
Direct recombinase-DBD fusions
The most direct approach to retargeting of viral or transposon vectors is to directly fuse a DBD to the recombinase enzyme (IN or transposase for virus- or transposon-based vector systems, respectively) (Figure 3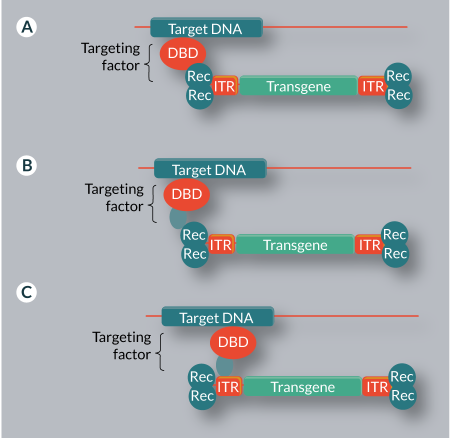
Several attempts have been made to retarget the HIV system by fusing DBDs to HIV IN. Fusions of HIV IN to LexA [115], lambda repressor (λR) [116] as well as the ZFPs Zif268 [117] and E2C [118] have been shown to bias HIV integration in cell-free in vitro assays using artificial target DNA fragments. The IN-E2C fusion was also shown to retarget HIV integration in cell culture assays [119]. In vitro, this construct increased the number of integrations in a 30-bp region around the target site six-fold [118], while in vivo, the percentage of insertions that occurred near the E2C binding site increased ten-fold [119]. All of these hybrid proteins retained catalytic activities similar to wild-type IN, but some resulted in reduced infectivity when assembled into virions [117]. Other IN fusions that have been successfully used to retarget retroviral systems include avian sarcoma virus (ASV) IN with LexA [120], and MLV IN with Sp1 [121].
Retargeting of the SB system via direct transposase fusions was first demonstrated by fusing the DBD of the yeast transcription factor Gal4 and the synthetic ZFP E2C to the N-terminus of the SB transposase [111]. While UAS, the target sequence of Gal4, is absent from the human genome, E2C targets a unique site near the erbB-2 gene [122,123]. These hybrid vectors were able to target integration near their respective target sites in inter-plasmid assays; integration near the target sites was increased up to 11-fold to 25% [111]. However, no targeting into the genome could be demonstrated. This was later achieved with the generation of new transposase hybrids: a fusion of E2C to a hyperactive SB mutant was shown to direct up to 2% of transposition events towards the endogenous target site in the promoter region of the erbB-2 gene [113], as demonstrated by a locus-specific semi-nested PCR assay. However, when the integration profile was analyzed on a whole-genome level by linear amplification mediated PCR, no bias toward integration near E2C recognition sites could be shown, suggesting that detection of these rare events was dependent on the method applied to recover the insertions. Additionally, a synthetic ZFP targeting the 3´-end of L1 elements was used to retarget the SB system [113]. L1 elements are good targets for SB transposition because they are A/T-rich (SB transposase integrates into TA dinucleotides) and abundant, making up 17% of the human genome [124]. The presence of multiple binding sites in the genome is expected to improve the ratio of targeted to untargeted events. In fact, around 45% of all transpositions catalyzed by the hybrid transposase ended up in L1 elements. A direct fusion of the DBD of the Rep protein from AAV to the SB transposase has also been shown to direct transposition towards endogenous RRSs [26].
Hybrid transposases based on the PB system have also been generated. The CHK2-ZFP, which targets the CHK2 gene with high specificity [125], can be fused to the PB transposase without significant loss of transpositional activity [126]. The hybrid vectors target a CHK2-ZFP binding site in an inter-plasmid transposition assay, but not the endogenous target site in the human genome [126]. However, a fusion of PB transposase and the Gal4 DBD was later shown to direct transposition to artificially introduced UAS sites as well as to endogenous UAS-like sites in the human genome (4.5-fold increase in an 800 bp window around endogenous sites) [110], and fusion with a synthetic TALE domain allowed targeting to the endogenous CCR5 gene (with 0.014% of cells containing targeted events) [109].
Other transposon systems that were retargeted using direct transposase-DBD fusions include the bacterial IS30 transposon (with cI repressor and Gli1) [127], the Drosophila Mos1 transposon (with Gal4) [128] and the bacterial ISY100 element (with the Zif268 ZFP) [129]. However, for all of these systems, targeted transposition was only demonstrated for integration into plasmids or bacterial genomes.
Transposases and viral INs are not the only recombinases that have been retargeted by fusing DBDs to them. While in tyrosine recombinases like Cre and FLP the catalytic domains and the DBDs are structurally intertwined [130], this is not true for serine recombinases. This family of enzymes, which contains the abovementioned ΦC31 IN, has physically separable DNA-binding and catalytic domains, making it possible to replace the DBD to alter the enzyme’s specificity [131]. Replacement of the original DBD with the Zif268 DBD has been shown to retarget Tn3 resolvase in bacterial cells [131]. Other serine recombinases, for example the Gin invertase, have also been fused to foreign DBDs and retargeting into the human genome has been demonstrated [132–134]. One such fusion protein consisted of the Gin catalytic domain and a catalytically inactivated Cas9 (dCas9) as a DBD, resulting in a recombinase that is active in human cells and can be targeted to extrachromosomal plasmids by supplying the proper gRNAs [135].
Targeting via adapter proteins
An alternative approach to vector retargeting, a mechanism found in most naturally targeted systems, is the use of adapter proteins that simultaneously bind to the target DNA and to a component of the vector system. This may be the integrating enzyme, the viral or transposon DNA, or both (Figure 3B–C & Table 3). This method avoids problems with activity loss associated with direct recombinase fusions.
| Table 3: Retargeting via adapter proteins. | ||||
|---|---|---|---|---|
| Adapter protein | Binding activities | Targeting effect | System | Ref. |
| Protein-chromatin adapters | ||||
| LEDGF/ λR | HIV-IN, λR site (genomic) | Increased integration near λR site | In vitro | [136] |
| LEDGF/CBX1 | HIV-IN, H3K9me3 | 50% decrease (from 67.2 to 32.6%) of insertions into genes | Cell culture (genomic) | [138] |
| LEDGF/ING2 | HIV-IN, H3K4me3 | 13-fold enrichment (from 3.8 to 50.3%) within 2.5 kbp of TSSs | Cell culture (genomic) | [140] |
| LEDGF/HP1α | HIV-IN, H3K9me2,3 | Ca. 1.5-fold enrichment in intergenic regions | Cell culture (genomic) | [141] |
| Sir4p/LexA | Ty5-IN, LexA site (genomic) | <200-fold enrichment of integration into target plasmid (from <0.1 to 15%) | Yeast (plasmid) | [81] |
| N57/TetR | SB transposase, TRE | >10% of all cells contained insertion near TRE | Cell culture (genomic) | [112] |
| N57/ZF-B | SB transposase, L1 elements | 4-fold enrichment within 400bp of ZF-B sites | Cell culture (genomic) | [113] |
| N57/E2C | SB transposase, erbB-2 locus | <1% insertion near erbB-2 locus | Cell culture (genomic) | [113] |
| N57/Rep | SB transposase, Rep recognition sequences | Ca. 2.5-fold enrichment near consensus RRSs | Cell culture (genomic) | [25] |
| DNA-chromatin adapters | ||||
| LexA/TetR | LexA site (SB transposon), TRE | <1% insertion near TRE | Cell culture (genomic) | [112] |
| LexA/SAF | LexA site (SB transposon), MARs | Ca. four-fold enrichment in MARs | Cell culture (genomic) | [112] |
| TALE/Gal4 | CCR5 locus, UAS (PB transposon) | 0.014% near CCR5 locus | Cell culture (genomic) | [109] |
Some retroviruses use this mechanism to direct their integration, and experimental manipulation of their targeting systems can alter their integration profile. LEDGF, a factor responsible for targeting of lentiviral integration, recognizes particular chromatin marks while simultaneously binding to HIV IN, thereby tethering the integration complexes to target sites in the genome. It is possible to retarget HIV integration by replacing the chromatin reading-domain of LEDGF with other DBDs. Fusing λR to the LEDGF IN-binding domain (IBD) targets HIV integration towards binding sites of λR in vitro [136]. Replacing the chromatin-binding domain of LEDGF with CBX1 (HP1β), which recognizes H3K9me3 chromatin marks [137], successfully retargeted HIV integration to intergenic regions in vivo [138]. While wild-type IN inserted into genes 67.2% of the time, this value dropped to 32.6% with the CBX1/LEDGF fusion [138]. This construct was even validated in an X-linked chronic granulomatous disease (X-CGD) model, demonstrating stable integration and expression [139]. Similar constructs with DBDs from ING2 and HP1α also altered the HIV integration profile in cell culture-based assays [140,141]. Deletion of the LEDGF PWWP domain or replacement with unspecific chromatin binding domains was also demonstrated to increase the percentage of ‘safe’ integrations [142].
An adapter-based approach has also been used to alter the target specificity of the Ty5 retrotransposon. A fusion of Sir4p, which interacts with Ty5 integrase during its natural targeting process, with LexA was used to direct insertions to a LexA binding site on a target plasmid [81]. Additionally, it was shown that target specificity could be altered by replacing the Sir4p interaction domain of the Ty5 IN [81].
Several adapter proteins have been developed for the SB system. For example, fusion proteins consisting of the LexA DBD and a second DBD with a genomic target were able to direct transposition of a transposon containing a LexA binding site [112]. Using the tetracycline repressor (TetR) as the second DBD allowed targeting of more than 10% of insertions towards an artificial tetracycline response element (TRE) containing binding sites for TetR, whereas a SAF-box fusion directed insertions to endogenous matrix attachment regions (MARs) [112].
Instead of using a DBD that binds to the transposon DNA, adapter proteins can also bind to the transposase. For this purpose, an N-terminal fragment of the SB transposase (N57), which is a dual DNA-binding and protein dimerization domain [143], can be used. Fusions of N57 with TetR, E2C, ZF-B and the Rep DBDs were able to direct transposition catalyzed by wild-type SB transposase to TRE, erbB-2 gene, L1 elements and RRSs, respectively [26,112,113]. Additionally, it has been shown that targeting efficiencies can be improved by utilizing fusion proteins that bind both the transposon DNA and the transposase [26].
So far, no equivalent to the N57 fragment is available for the PB system. Without a domain interacting with PB transposase, it is not possible to design a protein that would tether the transposase to the target site. However, it is possible to retarget PB transposition using adapter proteins consisting of two DBDs that bind to the target site and to a site in the transposon. This has been demonstrated by a fusion of a TALE domain recognizing the CCR5 locus in the human genome and a Gal4 domain in combination with a PB transposon containing a UAS site [109].
Translational Insight
The results of the studies mentioned above show that both viral and transposon vector systems can be retargeted using either direct recombinase fusions or adapter proteins. The efficiency of this targeting effect varies depending on the vector system used, the exact targeting mechanism and choice of target sites. However, for all of these approaches, the number of untargeted integrations is much higher than the number of targeted integrations. This is due to the fact that binding of the artificially introduced DBD is generally not required for the system to integrate. The number of potential integration sites is generally vastly greater than the number of desired integration sites, meaning that most integration events will occur bypassing the desired targeting effect. This problem could be addressed by modifying the vector system in a manner that makes integration dependent on binding of the foreign DBD. However, this has not been achieved yet.
Recent advances in the development of novel DBDs like ZFPs [144], TALEs [145] and the CRISPR/Cas system [146,147] have allowed the application of highly specific, designer endonucleases. Introduction of a double-strand break (DSB) at a precisely defined genomic location allows disruption of endogenous genes or – when coupled with a homology template – any desired modification of the target sequence [148], including gene repair and gene addition. In light of these developments, particularly the CRISPR/Cas system, it might seem that the development of targetable recombinases has become obsolete.
There are several important aspects to consider when comparing targeted viral/non-viral gene integration systems and designer nucleases. The first is the efficiency at which a desired genetic modification can be introduced into a cell population. Designer nucleases are specialized in introducing a DSB into the DNA, and are therefore highly efficient in mutagenizing a target site [149,150]. However, gene addition at the cut site is a process executed by DSB repair mechanisms of the cells; the efficiency of which is considerably lower than introducing the DSB in the first place [151]. In other words, knocking out a gene by designer nucleases is far more efficient than knocking in a gene into a specific site. On the other hand, integrating viruses and transposable elements have evolved machineries for gene integration, because genomic insertion is a fundamental step of the life cycle of these genetic elements. That means that the efficiency of gene insertion by vector systems that are based on such genetic elements is robust, which is a key requirement for medically relevant applications. An additional benefit of integrating vectors over nuclease-based approaches is that some integrating vectors, particularly those based on transposons, can deliver their cargo into the genomes of non-dividing cells [152,153]. Nuclease-based approaches, on the other hand, rely on DSB repair, as outlined above. In eukaryotic cells, DSBs can be repaired by at least two pathways, homology-directed repair (HDR) and non-homologous end joining (NHEJ). The two pathways act complementarily, but at different stages of the cell-cycle: NHEJ is preferentially active in the G1 and early S phases [154], whereas HDR is the preferentially used DSB repair pathway in the late S and G2 phases when homology templates are available [155], and is strongly downregulated in most post-mitotic cells [156]. Consequently, gene addition and gene repair require dividing target cells.
The second important aspect is the safety profiles of the diverse gene insertion technologies. Although gene insertion by designer nucleases is targeted to specific sites in the genome, cellular responses to DSBs can result in cytotoxic effects and off-target cleavage can lead to genotoxicity [157–162]. Due to its RNA-guided targeting mechanism, the CRISPR/Cas system is especially prone to off-target effects, which can be hard to predict. The extent of off-target effects varies with the actual gRNA sequence, but several mismatches can be tolerated, even if they occur consecutively [163]. In vitro, off-target sites with seven mismatches were observed [164] and modification of off-target sites can be as efficient or even more efficient than modification of the on-target site [163]. Genome-wide methods for off-target detection like GUIDE-seq [165] confirmed that the number of off-target sites and the efficiency at which they are modified strongly depends on the individual gRNAs. The number of off-target sites can range from none [165] to thousands [166], and bioinformatics tools like the MIT CRISPR Design Tool [167] or E-CRISP [168] might fail to predict experimentally determined off-target sites [165]. However, high-fidelity variants of Cas9 with greatly reduced off-target effects have been developed [169].
While the mechanism of HDR results in highly specific gene correction, any DSB can also be repaired by NHEJ. This means that, although cutting the DNA catalyzed by recombinases is highly specific, the actual outcome of the reaction on a genome-wide scale can be diverse. This has profound implications for the detection of off-target effects. It is relatively easy to determine the numbers and genomic locations of virus or transposon integrations in a target cell. Thus, picking cells with a single integration event mapped onto a genetic locus results in a high degree of certainty that: i) no other genomic modifications have been introduced; and ii) the insertion event has no negative impact on the cell as a risk factor. This is not possible when using endonuclease-based approaches because some off-target mutations – for example the deletion of a single nucleotide – are very difficult to detect. Thus, gene additions by integrating genetic vector systems (including the targeted recombinase approaches outlined in this article) could very well be safer than an endonuclease, in case clonal analysis of genetically engineered cell products is possible. In bulk cell populations, where prior annotation of every single insertion and off-target cleavage event is not feasible, potential risks associated with genome engineering can only be inferred on the basis of the type of vector used (insertional preferences of viral and non-viral vectors), the nuclease and the genomic target site (these will be the major determinants of off-target cleavage), the type of cell (stem cells with a high proliferative potential are more prone to oncogenic transformation) and the disease condition (which largely determines what cell types are to be engineered, and specifies a certain genetic background that might affect the risks associated with genetic engineering).
Financial & competing interests disclosure
The authors have no relevant financial involvement with an organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock options or ownership, expert testimony, grants or patents received or pending, or royalties. No writing assistance was utilized in the production of this manuscript.
References
1. Hacein-Bey-Abina S, Le Deist F, Carlier F et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N. Engl. J. Med. 2002; 346(16): 1185–93.
CrossRef
2. Herzog RW. Gene therapy for SCID-X1: round 2. Mol. Ther. 2010; 18(11): 1891.
CrossRef
3. Cavazzana M, Six E, Lagresle-Peyrou C, Andre-Schmutz I, Hacein-Bey-Abina S. Gene Therapy for X-Linked Severe Com-bined Immunodeficiency: Where Do We Stand? Hum. Gene Ther. 2016; 27(2): 108–16.
CrossRef
4. Hacein-Bey-Abina S, Kalle C von, Schmidt M et al. A serious adverse event after successful gene therapy for X-linked se-vere combined immunodeficiency. N. Engl. J. Med. 2003; 348(3): 255–6.
CrossRef
5. Hacein-Bey-Abina S, Kalle C von, Schmidt M et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003; 302(5644): 415–9.
CrossRef
6. Howe SJ, Mansour MR, Schwarzwaelder K et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest. 2008; 118(9): 3143–50.
CrossRef
7. Boztug K, Schmidt M, Schwarzer A et al. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N. Engl. J. Med. 2010; 363(20): 1918–27.
CrossRef
8. Stein S, Ott MG, Schultze-Strasser S et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010; 16(2): 198–204.
CrossRef
9. Touzot F, Moshous D, Creidy R et al. Faster T-cell development following gene therapy compared with haploidentical HSCT in the treatment of SCID-X1. Blood 2015; 125(23): 3563–9.
CrossRef
10. Kohn DB. Gene therapy outpaces haplo for SCID-X1. Blood 2015; 125(23): 3521–2.
CrossRef
11. Hacein-Bey-Abina S, Pai S-Y, Gaspar HB et al. A modified gamma-retrovirus vector for X-linked severe combined immu-nodeficiency. N. Engl. J. Med. 2014; 371(15): 1407–17.
CrossRef
12. Cartier N, Hacein-Bey-Abina S, Bartholomae CC et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009; 326(5954): 818–23.
CrossRef
13. Biffi A, Montini E, Lorioli L et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013; 341(6148): 1233158.
CrossRef
14. Levine BL, Humeau LM, Boyer J et al. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc. Natl Acad. Sci. USA 2006; 103(46): 17372–7.
CrossRef
15. Cavazzana-Calvo M, Payen E, Negre O et al. Transfusion independence and HMGA2 activation after gene therapy of hu-man beta-thalassaemia. Nature 2010; 467(7313): 318–22.
CrossRef
16. Hacein-Bey Abina S, Gaspar HB, Blondeau J et al. Outcomes following gene therapy in patients with severe Wiskott-Aldrich syndrome. JAMA 2015; 313(15): 1550–63.
CrossRef
17. Aiuti A, Biasco L, Scaramuzza S et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syn-drome. Science 2013; 341(6148): 1233151.
CrossRef
18. Brentjens RJ, Davila ML, Riviere I et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemo-therapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013; 5(177): 177ra38.
CrossRef
19. Kochenderfer JN, Dudley ME, Kassim SH et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015; 33(6): 540–9.
CrossRef
20. Lee DW, Kochenderfer JN, Stetler-Stevenson M et al. T cells expressing CD19 chimeric antigen receptors for acute lym-phoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015; 385(9967): 517–28.
CrossRef
21. Maude SL, Frey N, Shaw PA et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014; 371(16): 1507–17.
CrossRef
22. Schroder ARW, Shinn P, Chen H et al. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002; 110(4): 521–9.
CrossRef
23. Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science 2003; 300(5626): 1749–51.
CrossRef
24. Cavazza A, Moiani A, Mavilio F. Mechanisms of retroviral integration and mutagenesis. Hum. Gene Ther. 2013; 24(2): 119–31.
CrossRef
25. Gogol-Doring A, Ammar I, Gupta S et al. Genome-wide Profiling Reveals Remarkable Parallels Between Insertion Site Selection Properties of the MLV Retrovirus and the piggyBac Transposon in Primary Human CD4(+) T Cells. Mol. Ther. 2016; 24(3): 592–606.
CrossRef
26. Ammar I, Gogol-Doring A, Miskey C et al. Retargeting transposon insertions by the adeno-associated virus Rep protein. Nucleic Acids Res. 2012; 40(14): 6693–712.
CrossRef
27. Monjezi R, Miskey C, Gogishvili T et al. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia 2017; 31(1): 186–94.
CrossRef
28. Yant SR, Wu X, Huang Y et al. High-resolution genome-wide mapping of transposon integration in mammals. Mol. Cell Biol. 2005; 25(6): 2085–94.
CrossRef
29. Jong J de, Akhtar W, Badhai J et al. Chromatin landscapes of retroviral and transposon integration profiles. PLoS Genet. 2014; 10(4): e1004250.
CrossRef
30. Stocking C, Bergholz U, Friel J et al. Distinct classes of factor-independent mutants can be isolated after retroviral mutagen-esis of a human myeloid stem cell line. Growth Factors 1993; 8(3): 197–209.
CrossRef
31. Hacein-Bey-Abina S, Garrigue A, Wang GP et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene thera-py of SCID-X1. J. Clin. Invest. 2008; 118(9): 3132–42.
CrossRef
32. Ciuffi A, Llano M, Poeschla E et al. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005; 11(12): 1287–9.
CrossRef
33. Cherepanov P, Sun Z-YJ, Rahman S et al. Solution structure of the HIV-1 integrase-binding domain in LEDGF/p75. Nat. Struct. Mol. Biol. 2005; 12(6): 526–32.
CrossRef
34. Llano M, Saenz DT, Meehan A et al. An essential role for LEDGF/p75 in HIV integration. Science 2006; 314(5798): 461–4.
CrossRef
35. Busschots K, Vercammen J, Emiliani S et al. The interaction of LEDGF/p75 with integrase is lentivirus-specific and pro-motes DNA binding. J. Biol. Chem. 2005; 280(18): 17841–7.
CrossRef
36. Rijck J de, Bartholomeeusen K, Ceulemans H, Debyser Z, Gijsbers R. High-resolution profiling of the LEDGF/p75 chromatin interaction in the ENCODE region. Nucleic Acids Res. 2010; 38(18): 6135–47.
CrossRef
37. Eidahl JO, Crowe BL, North JA et al. Structural basis for high-affinity binding of LEDGF PWWP to mononucleosomes. Nucleic Acids Res. 2013; 41(6): 3924–36.
CrossRef
38. van Nuland R, van Schaik FM, Simonis M et al. Nucleosomal DNA binding drives the recognition of H3K36-methylated nucleosomes by the PSIP1-PWWP domain. Epigenetics Chromatin 2013; 6(1): 12.
CrossRef
39. Pradeepa MM, Sutherland HG, Ule J, Grimes GR, Bickmore WA. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012; 8(5): e1002717.
<a href=”https://doi.org/10.1371/journal.pgen.1002717
40. Singh PK, Plumb MR, Ferris AL et al. LEDGF/p75 interacts with mRNA splicing factors and targets HIV-1 integration to highly spliced genes. Genes Dev. 2015; 29(21): 2287–97.
CrossRef
41. Hombrouck A, Rijck J de, Hendrix J et al. Virus evolution reveals an exclusive role for LEDGF/p75 in chromosomal teth-ering of HIV. PLoS Pathog. 2007; 3(3): e47.
CrossRef
42. Cattoglio C, Pellin D, Rizzi E et al. High-definition mapping of retroviral integration sites identifies active regulatory ele-ments in human multipotent hematopoietic progenitors. Blood 2010; 116(25): 5507–17.
CrossRef
43. Mitchell RS, Beitzel BF, Schroder ARW et al. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004; 2(8): E234.
CrossRef
44. Sharma A, Larue RC, Plumb MR et al. BET proteins promote efficient murine leukemia virus integration at transcription start sites. Proc. Natl Acad. Sci. USA 2013; 110(29): 12036–41.
CrossRef
45. de Rijck J, de Kogel C, Demeulemeester J et al. The BET family of proteins targets moloney murine leukemia virus integra-tion near transcription start sites. Cell Rep. 2013; 5(4): 886–94.
CrossRef
46. Gupta SS, Maetzig T, Maertens GN et al. Bromo- and extraterminal domain chromatin regulators serve as cofactors for murine leukemia virus integration. J. Virol. 2013; 87(23): 12721–36.
CrossRef
47. Debyser Z, Christ F, Rijck J de, Gijsbers R. Host factors for retroviral integration site selection. Trends Biochem. Sci. 2015; 40(2): 108–16.
CrossRef
48. El Ashkar S, de Rijck J, Demeulemeester J et al. BET-independent MLV-based Vectors Target Away From Promoters and Regulatory Elements. Mol. Ther. Nucleic Acids 2014; 3: e179.
CrossRef
49. Kotin RM, Siniscalco M, Samulski RJ et al. Site-specific integration by adeno-associated virus. Proc. Natl Acad. Sci. USA 1990; 87(6): 2211–5.
CrossRef
50. Giraud C, Winocour E, Berns KI. Site-specific integration by adeno-associated virus is directed by a cellular DNA se-quence. Proc. Natl Acad. Sci. USA 1994; 91(21): 10039–43.
CrossRef
51. Henckaerts E, Dutheil N, Zeltner N et al. Site-specific integration of adeno-associated virus involves partial duplication of the target locus. Proc. Natl Acad. Sci. USA 2009; 106(18): 7571–6.
CrossRef
52. McCarty DM, Ryan JH, Zolotukhin S, Zhou X, Muzyczka N. Interaction of the adeno-associated virus Rep protein with a sequence within the A palindrome of the viral terminal repeat. J. Virol. 1994; 68(8): 4998–5006.
53. Daya S, Berns KI. Gene therapy using adeno-associated virus vectors. Clin. Microbiol. Rev. 2008; 21(4): 583–93.
CrossRef
54. Goncalves, Manuel AFV. Adeno-associated virus: from defective virus to effective vector. Virol J. 2005; 2: 43.
CrossRef
55. Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat. Rev. Genet. 2011; 12(5): 341–55.
CrossRef
56. Skipper KA, Andersen PR, Sharma N, Mikkelsen JG. DNA transposon-based gene vehicles – scenes from an evolutionary drive. J. Biomed. Sci. 2013; 20: 92.
CrossRef
57. Voigt K, Izsvak Z, Ivics Z. Targeted gene insertion for molecular medicine. J. Mol. Med. (Berl.). 2008; 86(11): 1205–19.
CrossRef
58. Chalker DL, Sandmeyer SB. Ty3 integrates within the region of RNA polymerase III transcription initiation. Genes Dev. 1992; 6(1): 117–28.
CrossRef
59. Devine SE, Boeke JD. Integration of the yeast retrotransposon Ty1 is targeted to regions upstream of genes transcribed by RNA polymerase III. Genes Dev. 1996; 10(5): 620–33.
CrossRef
60. Boeke JD, Devine SE. Yeast retrotransposons: finding a nice quiet neighborhood. Cell 1998; 93(7): 1087–9.
CrossRef
61. Ji H, Moore DP, Blomberg MA et al. Hotspots for unselected Ty1 transposition events on yeast chromosome III are near tRNA genes and LTR sequences. Cell 1993; 73(5): 1007–18.
CrossRef
62. Kim JM, Vanguri S, Boeke JD, Gabriel A, Voytas DF. Transposable elements and genome organization: a comprehensive survey of retrotransposons revealed by the complete Saccharomyces cerevisiae genome sequence. Genome Res. 1998; 8(5): 464–78.
63. Bryk M, Banerjee M, Murphy M et al. Transcriptional silencing of Ty1 elements in the RDN1 locus of yeast. Genes Dev. 1997; 11(2): 255–69.
CrossRef
64. Bachman N, Gelbart ME, Tsukiyama T, Boeke JD. TFIIIB subunit Bdp1p is required for periodic integration of the Ty1 retrotransposon and targeting of Isw2p to S. cerevisiae tDNAs. Genes Dev. 2005; 19(8): 955–64.
CrossRef
65. Curcio MJ, Lutz S, Lesage P. The Ty1 LTR-Retrotransposon of Budding Yeast, Saccharomyces cerevisiae. Microbiol. Spectr. 2015; 3(2): MDNA3-0053-2014.
66. Aye M, Dildine SL, Claypool JA, Jourdain S, Sandmeyer SB. A truncation mutant of the 95-kilodalton subunit of transcrip-tion factor IIIC reveals asymmetry in Ty3 integration. Mol. Cell Biol. 2001; 21(22): 7839–51.
CrossRef
67. Kirchner J, Connolly CM, Sandmeyer SB. Requirement of RNA polymerase III transcription factors for in vitro position-specific integration of a retroviruslike element. Science 1995; 267(5203): 1488–91.
CrossRef
68. Yieh L, Hatzis H, Kassavetis G, Sandmeyer SB. Mutational analysis of the transcription factor IIIB-DNA target of Ty3 retroelement integration. J. Biol. Chem. 2002; 277(29): 25920–8.
CrossRef
69. Chung T, Siol O, Dingermann T, Winckler T. Protein interactions involved in tRNA gene-specific integration of Dictyostelium discoideum non-long terminal repeat retrotransposon TRE5-A. Mol. Cell Biol. 2007; 27(24): 8492–501.
CrossRef
70. Winckler T, Dingermann T, Glockner G. Dictyostelium mobile elements: strategies to amplify in a compact genome. Cell Mol. Life Sci. 2002; 59(12): 2097–111.
CrossRef
71. Winckler T, Szafranski K, Glockner G. Transfer RNA gene-targeted integration: an adaptation of retrotransposable ele-ments to survive in the compact Dictyostelium discoideum genome. Cytogenet. Genome Res. 2005; 110(1–4): 288–98.
CrossRef
72. Guo Y, Levin HL. High-throughput sequencing of retrotransposon integration provides a saturated profile of target activi-ty in Schizosaccharomyces pombe. Genome Res. 2010; 20(2): 239–48.
CrossRef
73. Chatterjee AG, Esnault C, Guo Y et al. Serial number tagging reveals a prominent sequence preference of retrotransposon integration. Nucleic Acids Res. 2014; 42(13): 8449–60.
CrossRef
74. Arcangioli B, Ghazvini M, Ribes V. Identification of the DNA-binding domains of the switch-activating-protein Sap1 from S.pombe by random point mutations screening in E. coli. Nucleic Acids Res. 1994; 22(15): 2930–7.
CrossRef
75. Jacobs JZ, Rosado-Lugo JD, Cranz-Mileva S et al. Arrested replication forks guide retrotransposon integration. Science 2015; 349(6255): 1549–53.
CrossRef
76. Zou S, Ke N, Kim JM, Voytas DF. The Saccharomyces retrotransposon Ty5 integrates preferentially into regions of silent chromatin at the telomeres and mating loci. Genes Dev. 1996; 10(5): 634–45.
CrossRef
77. Zou S, Kim JM, Voytas DF. The Saccharomyces retrotransposon Ty5 influences the organization of chromosome ends. Nucleic Acids Res. 1996; 24(23): 4825–31.
CrossRef
78. Zou S, Voytas DF. Silent chromatin determines target preference of the Saccharomyces retrotransposon Ty5. Proc. Natl Acad. Sci. USA 1997; 94(14): 7412–6.
CrossRef
79. Gai X, Voytas DF. A single amino acid change in the yeast retrotransposon Ty5 abolishes targeting to silent chromatin. Mol. Cell. 1998; 1(7): 1051–5.
CrossRef
80. Xie W, Gai X, Zhu Y et al. Targeting of the yeast Ty5 retrotransposon to silent chromatin is mediated by interactions be-tween integrase and Sir4p. Mol. Cell Biol. 2001; 21(19): 6606–14.
CrossRef
81. Zhu Y, Dai J, Fuerst PG, Voytas DF. Controlling integration specificity of a yeast retrotransposon. Proc. Natl Acad. Sci. USA 2003; 100(10): 5891–5.
CrossRef
82. Cockell M, Palladino F, Laroche T et al. The carboxy termini of Sir4 and Rap1 affect Sir3 localization: evidence for a multicomponent complex required for yeast telomeric silencing. J. Cell Biol. 1995; 129(4): 909–24.
CrossRef
83. Palladino F, Laroche T, Gilson E et al. SIR3 and SIR4 proteins are required for the positioning and integrity of yeast telomeres. Cell 1993; 75(3): 543–55.
CrossRef
84. Peters JE, Craig NL. Tn7 recognizes transposition target structures associated with DNA replication using the DNA-binding protein TnsE. Genes Dev. 2001; 15(6): 737–47.
CrossRef
85. Parks AR, Li Z, Shi Q et al. Transposition into replicating DNA occurs through interaction with the processivity factor. Cell 2009; 138(4): 685–95.
CrossRef
86. Bainton RJ, Kubo KM, Feng JN, Craig NL. Tn7 transposition: target DNA recognition is mediated by multiple Tn7-encoded proteins in a purified in vitro system. Cell 1993; 72(6): 931–43.
CrossRef
87. Gringauz E, Orle KA, Waddell CS, Craig NL. Recognition of Escherichia coli attTn7 by transposon Tn7: lack of specific sequence requirements at the point of Tn7 insertion. J. Bacteriol. 1988; 170(6): 2832–40.
CrossRef
88. Peters JE, Craig NL. Tn7: smarter than we thought. Nat. Rev. Mol. Cell Biol. 2001; 2(11): 806–14.
CrossRef
89. Kuduvalli PN, Mitra R, Craig NL. Site-specific Tn7 transposition into the human genome. Nucleic Acids Res. 2005; 33(3): 857–63.
CrossRef
90. Thorpe HM, Smith MC. In vitro site-specific integration of bacteriophage DNA catalyzed by a recombinase of the resolvase/invertase family. Proc. Natl Acad. Sci. USA 1998; 95(10): 5505–10.
CrossRef
91. Groth AC, Olivares EC, Thyagarajan B, Calos MP. A phage integrase directs efficient site-specific integration in human cells. Proc. Natl Acad. Sci. USA 2000; 97(11): 5995–6000.
CrossRef
92. Thyagarajan B, Olivares EC, Hollis RP, Ginsburg DS, Calos MP. Site-Specific Genomic Integration in Mammalian Cells Mediated by Phage C31 Integrase. Mol. Cell Biol. 2001; 21(12): 3926–34.
CrossRef
93. Chavez CL, Calos MP. Therapeutic applications of the PhiC31 integrase system. Curr. Gene Ther. 2011; 11(5): 375–81.
CrossRef
94. Olivares EC, Hollis RP, Chalberg TW et al. Site-specific genomic integration produces therapeutic Factor IX levels in mice. Nat. Biotechnol. 2002; 20(11): 1124–8.
CrossRef
95. Ortiz-Urda S, Thyagarajan B, Keene DR et al. Stable nonviral genetic correction of inherited human skin disease. Nat. Med. 2002; 8(10): 1166–70.
CrossRef
96. Ehrhardt A, Xu H, Huang Z, Engler JA, Kay MA. A direct comparison of two nonviral gene therapy vectors for somatic integration: in vivo evaluation of the bacteriophage integrase phiC31 and the Sleeping Beauty transposase. Mol. Ther. 2005; 11(5): 695–706.
CrossRef
97. Held PK, Olivares EC, Aguilar CP et al. In vivo correction of murine hereditary tyrosinemia type I by phiC31 integrase-mediated gene delivery. Mol. Ther. 2005; 11(3): 399–408.
CrossRef
98. Chalberg TW, Genise HL, Vollrath D, Calos MP. phiC31 integrase confers genomic integration and long-term transgene expression in rat retina. Invest. Ophthalmol. Vis. Sci. 2005; 46(6): 2140–6.
CrossRef
99. Liu J, Skjorringe T, Gjetting T, Jensen TG. PhiC31 integrase induces a DNA damage response and chromosomal rearrangements in human adult fibroblasts. BMC Biotechnol. 2009; 9: 31
CrossRef
100. Liu J, Jeppesen I, Nielsen K, Jensen TG. Phi c31 integrase induces chromosomal aberrations in primary human fibroblasts. Gene Ther. 2006; 13(15): 1188–90.
CrossRef
101. Ivics Z, Hackett PB, Plasterk RH, Izsvak Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 1997; 91(4): 501–10.
CrossRef
102. Brady TL, Schmidt CL, Voytas DF. Targeting integration of the Saccharomyces Ty5 retrotransposon. Methods Mol Biol. 2008; 435: 153–63.
CrossRef
103. Wang H, Mayhew D, Chen X, Johnston M, Mitra RD. “Calling cards” for DNA-binding proteins in mammalian cells. Genetics 2012; 190(3) :941–9.
CrossRef
104. Wang H, Mayhew D, Chen X, Johnston M, Mitra RD. Calling Cards enable multiplexed identification of the genomic targets of DNA-binding proteins. Genome Res. 2011; 21(5): 748–55.
CrossRef
105. Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev. Genet. 2010; 11(9): 636–46.
CrossRef
106. Ousterout DG, Gersbach CA. The Development of TALE Nucleases for Biotechnology. Methods Mol. Biol. 2016; 1338: 27–42.
CrossRef
107. Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014; 346(6213): 1258096.
CrossRef
108. Wu SC-Y, Meir Y-JJ, Coates CJ et al. piggyBac is a flexible and highly active transposon as compared to sleeping beauty, Tol2, and Mos1 in mammalian cells. Proc. Natl Acad Sci. USA 2006; 103(41): 15008–13.
CrossRef
109. Owens JB, Mauro D, Stoytchev I et al. Transcription activator like effector (TALE)-directed piggyBac transposition in human cells. Nucleic Acids Res. 2013; 41(19): 9197–207.
CrossRef
110. Owens JB, Urschitz J, Stoytchev I et al. Chimeric piggyBac transposases for genomic targeting in human cells. Nucleic Acids Res. 2012; 40(14): 6978–91.
CrossRef
111. Yant SR, Huang Y, Akache B, Kay MA. Site-directed transposon integration in human cells. Nucleic Acids Res. 2007; 35(7): e50.
CrossRef
112. Ivics Z, Katzer A, Stuwe EE et al. Targeted Sleeping Beauty transposition in human cells. Mol. Ther. 2007; 15(6): 1137–44.
CrossRef
113. Voigt K, Gogol-Doring A, Miskey C et al. Retargeting sleeping beauty transposon insertions by engineered zinc finger DNA-binding domains. Mol. Ther. 2012; 20(10): 1852–62.
CrossRef
114. Wilson MH, Kaminski JM, George AL, JR. Functional zinc finger/sleeping beauty transposase chimeras exhibit attenuated overproduction inhibition. FEBS Lett. 2005; 579(27): 6205–9.
CrossRef
115. Goulaouic H, Chow SA. Directed integration of viral DNA mediated by fusion proteins consisting of human immunodeficiency virus type 1 integrase and Escherichia coli LexA protein. J. Virol. 1996; 70(1): 37–46.
116. Bushman FD. Tethering human immunodeficiency virus 1 integrase to a DNA site directs integration to nearby sequences. Proc. Natl Acad. Sci. USA 1994; 91(20): 9233–7.
CrossRef
117. Bushman FD, Miller MD. Tethering human immunodeficiency virus type 1 preintegration complexes to target DNA promotes integration at nearby sites. J. Virol. 1997; 71(1): 458–64.
118. Tan W, Zhu K, Segal DJ, Barbas CF, Chow SA. Fusion Proteins Consisting of Human Immunodeficiency Virus Type 1 Integrase and the Designed Polydactyl Zinc Finger Protein E2C Direct Integration of Viral DNA into Specific Sites. J. Virol. 2004; 78(3): 1301–13.
CrossRef
119. Tan W, Dong Z, Wilkinson TA, Barbas CF3, Chow SA. Human immunodeficiency virus type 1 incorporated with fusion proteins consisting of integrase and the designed polydactyl zinc finger protein E2C can bias integration of viral DNA into a predetermined chromosomal region in human cells. J. Virol. 2006; 80(4): 1939–48.
CrossRef
120. Katz RA, Merkel G, Skalka AM. Targeting of retroviral integrase by fusion to a heterologous DNA binding domain: in vitro activities and incorporation of a fusion protein into viral particles. Virology 1996; 217(1): 178–90.
<a href=”https://doi.org/10.1006/viro.1996.0105
121. Peng W-J, Chang C-M, Lin T-H. Target integration by a chimeric Sp1 zinc finger domain-Moloney murine leukemia virus integrase in vivo. J. Biomed. Sci. 2002; 9(2): 171–84.
CrossRef
122. Beerli RR, Dreier B, Barbas CF3. Positive and negative regulation of endogenous genes by designed transcription factors. Proc. Natl Acad. Sci. USA 2000; 97(4): 1495–500.
CrossRef
123. Beerli RR, Segal DJ, Dreier B, Barbas CF3. Toward controlling gene expression at will: specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc. Natl Acad. Sci. USA 1998; 95(25): 14628–33.
CrossRef
124. Lander ES, Linton LM, Birren B et al. Initial sequencing and analysis of the human genome. Nature 2001; 409(6822): 860–921.
CrossRef
125. Tan S, Guschin D, Davalos A et al. Zinc-finger protein-targeted gene regulation: genomewide single-gene specificity. Proc. Natl Acad. Sci. USA 2003; 100(21): 11997–2002.
CrossRef
126. Kettlun C, Galvan DL, George AL, JR, Kaja A, Wilson MH. Manipulating piggyBac transposon chromosomal integration site selection in human cells. Mol. Ther. 2011; 19(9): 1636–44.
CrossRef
127. Szabo M, Muller F, Kiss J, Balduf C, Strahle U, Olasz F. Transposition and targeting of the prokaryotic mobile element IS30 in zebrafish. FEBS Lett. 2003; 550(1–3): 46–50.
CrossRef
128. Maragathavally KJ, Kaminski JM, Coates CJ. Chimeric Mos1 and piggyBac transposases result in site-directed integration. FASEB J. 2006; 20(11): 1880–2.
CrossRef
129. Feng X, Bednarz AL, Colloms SD. Precise targeted integration by a chimaeric transposase zinc-finger fusion protein. Nucleic Acids Res. 2010; 38(4): 1204–16.
CrossRef
130. Guo F, Gopaul DN, van Duyne GD. Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature 1997; 389(6646): 40–6.
CrossRef
131. Akopian A, He J, Boocock MR, Stark WM. Chimeric recombinases with designed DNA sequence recognition. Proc. Natl Acad. Sci. USA 2003; 100(15): 8688–91.
CrossRef
132. Gordley RM, Smith JD, Graslund T, Barbas CF3. Evolution of programmable zinc finger-recombinases with activity in human cells. J. Mol. Biol. 2007; 367(3): 802–13.
CrossRef
133. Gordley RM, Gersbach CA, Barbas CF3. Synthesis of programmable integrases. Proc. Natl Acad. Sci. USA 2009; 106(13): 5053–8.
CrossRef
134. Schneider F, Schwikardi M, Muskhelishvili G, Droge P. A DNA-binding domain swap converts the invertase gin into a resolvase. J. Mol. Biol. 2000; 295(4): 767–75.
CrossRef
135. Chaikind B, Bessen JL, Thompson DB, Hu JH, Liu DR. A programmable Cas9-serine recombinase fusion protein that operates on DNA sequences in mammalian cells. Nucleic Acids Res. 2016; 44(20): 9758–70.
CrossRef
136. Ciuffi A, Diamond TL, Hwang Y, Marshall HM, Bushman FD. Modulating target site selection during human immunodeficiency virus DNA integration in vitro with an engineered tethering factor. Hum. Gene Ther. 2006; 17(9): 960–7.
CrossRef
137. Dialynas GK, Makatsori D, Kourmouli N et al. Methylation-independent binding to histone H3 and cell cycle-dependent incorporation of HP1beta into heterochromatin. J. Biol. Chem. 2006; 281(20): 14350–60.
CrossRef
138. Gijsbers R, Ronen K, Vets S et al. LEDGF hybrids efficiently retarget lentiviral integration into heterochromatin. Mol. Ther. 2010; 18(3): 552–60.
CrossRef
139. Vets S, Rijck J de, Brendel C et al. Transient Expression of an LEDGF/p75 Chimera Retargets Lentivector Integration and Functionally Rescues in a Model for X-CGD. Mol. Ther. Nucleic Acids 2013; 2: e77.
CrossRef
140. Ferris AL, Wu X, Hughes CM et al. Lens epithelium-derived growth factor fusion proteins redirect HIV-1 DNA integration. Proc. Natl Acad. Sci. USA 2010; 107(7): 3135–40.
CrossRef
141. Silvers RM, Smith JA, Schowalter M et al. Modification of integration site preferences of an HIV-1-based vector by expression of a novel synthetic protein. Hum. Gene Ther. 2010; 21(3): 337–49.
CrossRef
142. Vranckx LS, Demeulemeester J, Debyser Z, Gijsbers R. Towards a Safer, More Randomized Lentiviral Vector Integration Profile Exploring Artificial LEDGF Chimeras. PLoS One 2016; 11(10): e0164167.
CrossRef
143. Izsvak Z, Khare D, Behlke J, Heinemann U, Plasterk RH, Ivics Z. Involvement of a bifunctional, paired-like DNA-binding domain and a transpositional enhancer in Sleeping Beauty transposition. J. Biol. Chem. 2002; 277(37): 34581–8.
CrossRef
144. Perez EE, Wang J, Miller JC et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 2008; 26(7): 808–16.
CrossRef
145. Li T, Huang S, Jiang WZ et al. TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Res. 2011; 39(1): 359–72.
CrossRef
146. Le Cong, Ran FA, Cox D et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013; 339(6121): 819–23.
CrossRef
147. Mali P, Yang L, Esvelt KM et al. RNA-guided human genome engineering via Cas9. Science 2013; 339(6121): 823–6.
CrossRef
148. Porteus MH, Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science 2003; 300(5620): 763.
CrossRef
149. Hockemeyer D, Soldner F, Beard C et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009; 27(9): 851–7.
CrossRef
150. Hockemeyer D, Wang H, Kiani S et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 2011; 29(8): 731–4.
CrossRef
151. Zou J, Maeder ML, Mali P et al. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell. 2009; 5(1): 97–110.
CrossRef
152. Yant SR, Meuse L, Chiu W, Ivics Z, Izsvak Z, Kay MA. Somatic integration and long-term transgene expression in normal and haemophilic mice using a DNA transposon system. Nat. Genet. 2000; 25(1): 35–41.
CrossRef
153. Walisko O, Izsvak Z, Szabo K, Kaufman CD, Herold S, Ivics Z. Sleeping Beauty transposase modulates cell-cycle progression through interaction with Miz-1. Proc. Natl Acad. Sci. USA 2006; 103(11): 4062–7.
CrossRef
154. Lee SE, Mitchell RA, Cheng A, Hendrickson EA. Evidence for DNA-PK-dependent and -independent DNA double-strand break repair pathways in mammalian cells as a function of the cell cycle. Mol. Cell Biol. 1997; 17(3): 1425–33.
CrossRef
155. Takata M, Sasaki MS, Sonoda E et al. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998; 17(18): 5497–508.
CrossRef
156. Fung H, Weinstock DM. Repair at single targeted DNA double-strand breaks in pluripotent and differentiated human cells. PLoS One 2011; 6(5): e20514.
CrossRef
157. Bohne J, Cathomen T. Genotoxicity in gene therapy: an account of vector integration and designer nucleases. Curr. Opin. Mol. Ther. 2008; 10(3): 214–23.
158. Cornu TI, Cathomen T. Quantification of zinc finger nuclease-associated toxicity. Methods Mol. Biol. 2010; 649: 237–45.
CrossRef
159. Cornu TI, Thibodeau-Beganny S, Guhl E et al. DNA-binding specificity is a major determinant of the activity and toxicity of zinc-finger nucleases. Mol. Ther. 2008; 16(2): 352–8.
CrossRef
160. Gupta A, Meng X, Zhu LJ, Lawson ND, Wolfe SA. Zinc finger protein-dependent and -independent contributions to the in vivo off-target activity of zinc finger nucleases. Nucleic Acids Res. 2011; 39(1): 381–92.
CrossRef
161. Radecke S, Radecke F, Cathomen T, Schwarz K. Zinc-finger nuclease-induced gene repair with oligodeoxynucleotides: wanted and unwanted target locus modifications. Mol. Ther. 2010; 18(4): 743–53.
CrossRef
162. Pattanayak V, Ramirez CL, Joung JK, Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat. Methods 2011; 8(9): 765–70.
CrossRef
163. Fu Y, Foden JA, Khayter C et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013; 31(9): 822–6.
CrossRef
164. Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013; 31(9): 839–43.
CrossRef
165. Tsai SQ, Zheng Z, Nguyen NT et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015; 33(2): 187–97.
CrossRef
166. Kim D, Bae S, Park J et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods 2015; 12(3): 237-43.
CrossRef
167. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013; 8(11): 2281–308.
CrossRef
168. Heigwer F, Kerr G, Boutros M. E-CRISP: fast CRISPR target site identification. Nat. Methods 2014; 11(2): 122–3.
CrossRef
169. Kleinstiver BP, Pattanayak V, Prew MS et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016; 529(7587): 490–5.
CrossRef
Affiliations
Adrian Kovač and Zoltán Ivics*
Transposition and Genome Engineering, Division of Medical Biotechnology, Paul Ehrlich Institute, Langen, Germany.
* Author for correspondence: zoltan.ivics@pei.de

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.