Current gene editing strategies for the mucopolysaccharidoses
Cell Gene Therapy Insights 2017; 3(1), 17-31.
10.18609/cgti.2017.001
The mucopolysaccharidoses are one of the subclassifications of a group of metabolic disorders termed lysosomal storage disease. They occur from mutations to genes encoding enzymes responsible for glycosaminoglycan degradation. The ramifications of glycosaminoglycan accumulation are far reaching and result in pathology in multiple organ systems. Because storage diseases are monogenic disorders they are ideal candidates for gene and cell therapy. Importantly, cells distant from the site of enzyme production can benefit from secreted enzyme. Strategies for functionally restoring gene expression include gene therapy and gene editing. The latter employs tailored reagents (zinc finger nucleases, meganucleases, transcription activator-like effector nucleases and clustered regularly interspaced short palindromic repeats/Cas9) capable of generating DNA breaks at user-defined sites. Break resolution by homologous recombination can result in sequence modification for proper, or enhanced, gene expression. This powerful approach holds tremendous promise and here we discuss the employment of programmable nucleases to achieve therapeutic levels of enzyme for treating the pathogenic spectrum of mucopolysaccharidoses.
In 1955, Dr Christian de Duve described granules that contained hydrolytic enzymes that were distinct from mitochondria and microsomes that he termed lysosomes [1,2]. This organelle is comprised of membrane proteins responsible for the integrity of the vesicle in which approximately 60 hydrolases operate in mediating the stepwise degradation of macromolecules. The disruption of lysosomal function from a lack of a lysosomal hydrolase, the protein involved in hydrolase activation, or proper lysosomal peptide trafficking and transport, causes the accumulation of partially degraded substrates (Figure 1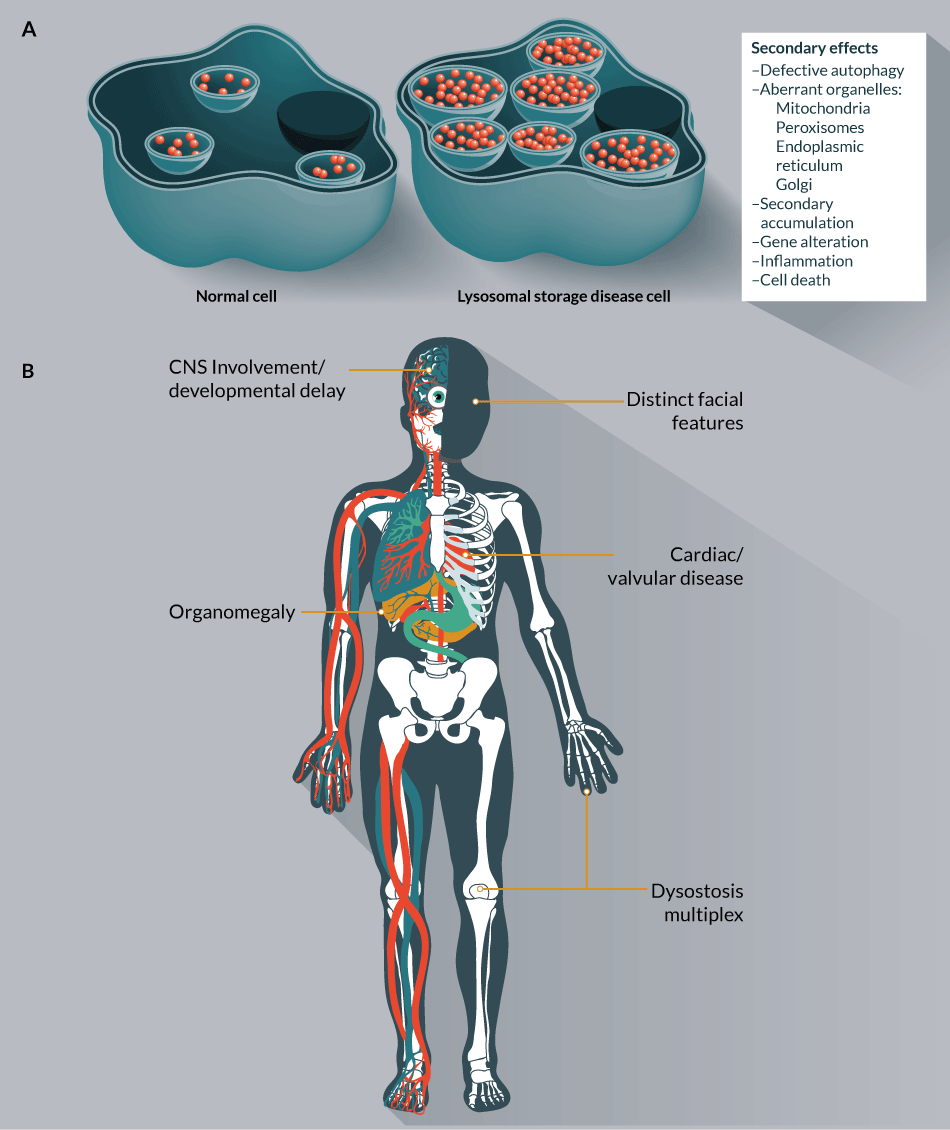
The MPS will be the focus of this review and are classified into seven groups (Table 1). The frequency of MPS can be variable with geography and individually are considered rare diseases; however, as a collective group their presence can be common with an occurrence of 1:5000 [5–9]. The inheritance pattern, save for the X-linked Hunter syndrome (MPS II), is autosomal recessive and the storage material in MPS disease is glycosaminoglycans (GAG) (Table 1). There are four types of GAGs that are linear, negatively charged carbohydrates comprised of repeating disaccharide units: heparan/heparin sulphate, chondroitin/dermatan sulphate, keratan sulphate and hyaluronic acid [10]. Collectively, GAGs interact with a wide variety of proteins and therefore are involved as initiators, regulators and orchestrators at both the cellular and organ level in diverse processes such as metabolism, transport, regulation and structural support [11]. The ramifications of gene mutations to lysosomal enzymes with subsequent accumulation of GAGs can be far reaching. Severe forms of MPS have multiorgan system pathology involving the skeletal system (dysostosis multiplex), organomegaly, respiratory and vision problems, neurological involvement and cardiac symptoms [12]. Attenuated forms of the disease can have less severe involvement but common to all MPS is some form of skeletal manifestation, coarse facial features and organomegaly [12,13].
| TABLE 1: Shown are the named disorders classified as MPSs. | |||
|---|---|---|---|
| Disease | Gene | ORF size (bp) | Storage material |
| MPS I (Hurler, Hurler-Scheie, Scheie) | IDUA | 1962 | Heparan and Dermatan sulphate |
| MPS II (Hunter) | IDS | 1653 | Heparan and Dermatan sulphate |
| MPS IIIA (Sanfilippo) | SGSH | 1509 | Heparan sulphate |
| MPS IIIB | NAGLU | 2232 | Heparan sulphate |
| MPS IIIC | HGSNAT | 1908 | Heparan sulphate |
| MPS IIID | GNS | 1659 | Heparan sulphate |
| MPS IV (Morquio-A) | GALNS | 1569 | Keratan sulphate, chondroitin-6- sulphate |
| MPS IV (Morquio-B) | GLB1 | 2034 | Keratan sulphate |
| MPS VI (Maroteaux-Lamy) | ASRB | 1602 | Dermatan sulphate |
| MPS VII (Sly) | GUSB | 1956 | Heparan and Dermatan sulphate and chondroitin-4- and 6- sulphate |
| MPS IX | HYAL1 | 1308 | Hyaluronan |
| The MPS disease class, gene responsible, size of the open reading frame and substrate that accumulates due to gene inactivation are shown. ORF=open reading frame. | |||
Given the role GAGs play in a broad number of biological processes from embryogenesis throughout the lifespan of an organism it is likely that failure to catabolize GAGs leads to dysregulation of a multitude of events. Initially, the cytotoxicity hypothesis put forth by Dr Robert Desnik stated that the loss of a lysosomal enzyme results in the build-up of non-degraded substrate causing endosomal/lysosomal (E/L) constipation, leading to cellular stress and death [14]. Dr Steven Walkley expands this notion to further characterize lysosomal storage diseases as states of deficiency [15]. Rather than a simple overabundance of storage material, this explanation suggests that the failure of the E/L system to recycle essential materials results in a depletion of metabolite building blocks [15]. In order to resupply itself with these precursors, the cell undertakes compensatory measures such as alternative regulation of synthesis pathways and/or induction of autophagy [15]. The role of autophagy in LSD pathophysiology is gaining prominence as a driver of secondary effects. Because of the diminished ability of GAG distended lysosomes to fuse with autophagosomes, there is a block in autophagic flux [16,17]. This term refers to the process of autophagosome formation, delivery to and degradation by the lysosome [18]. The pathological cascade radiates to other organelles including the Golgi, endoplasmic reticulum (ER), mitochondria and peroxisomes [19,20]. Golgi dysfunction with altered structure and impaired cellular trafficking has been observed in MPS IIIB mice [21]. Increased calcium stores in the ER of MPS cells has been demonstrated [22] as has the increased abundance of the Pdia5 gene [23], which is implicated as a marker of ER stress [24,25]. In MPS IIIC mice, the mitochondria of neurons showed impaired energy metabolism and altered structure [26]. Mitochondrial involvement with oxidative damage has also been observed [27,28]. Peroxisomal dysfunction occurs in storage disorders such as Krabbe [29] and Niemann Pick [30] as well as peroxisomal biogenesis disorders due to ganglioside accumulation [31]. Given that secondary ganglioside accumulation [32] is observed in MPS the subcellular impact of accumulated GAGs may extend to the perioxisome as well.
Collectively, the lysosome is a regulatory hub [33] and critical command and control [34] system involved in secretion, waste clearance, immune responses, gene regulation and integration of differential signals into a specified action of the cell. Thus, the disruption of lysosomal homeostasis via an inactivating mutation to a lysosomal hydrolase gene triggers a cascade of secondary events that cause/contribute to the complex and far reaching pathological manifestations (Figure 1B). The amount of storage and each tissue’s ability to tolerate perturbations in processes regulated by GAGs and the integrative lysosomal network as well as its capacity for regeneration following damage also likely play a role in the severity of organ level pathology. Both the initial genetic lesion causing GAG accumulation and the subsequent secondary cascade of downstream pathological events have implications for therapeutic intervention.
In 1968, work performed in the laboratory of Dr Elizabeth Neufeld described the ‘cross correction’ model that forms the basis of LSD treatment. Her group showed that extracellular enzyme can be endocytosed by the mannose 6-phosphate receptor (M6P) on the cell surface and routed to the lysosome where it can act upon accumulated GAGs [35,36]. This groundbreaking work showed that exogenous sources of enzyme could provide therapeutic benefit and that this can occur via a ‘bystander’ effect whereby the enzyme is provided/produced at sites distant to the cells that mediate uptake. Sources of enzymes for clinical and experimental treatment to date include recombinant enzyme infusion, delivery of allogeneic donor or gene modified autologous cells, infusion of gene therapy vehicles and gene editing.
Recombinant enzyme replacement therapy (ERT) relies on repetitive injections of purified enzyme. While effective, particularly for visceral pathological manifestations, the weekly injections are associated with significant cost and the potential for antibody formation. A more durable, lifelong source of enzyme has been cellular therapy with enzyme-producing cells.
In 1981, Dr JR Hobbs and colleagues performed the first allogeneic bone marrow transplant (alloBMT) for a patient with Hurler syndrome (MPS IH) [37]. In this strategy, the source of lysosomal enzyme was/is donor-derived stem cells that reconstitute the hematopoietic system and provide a continuous source of therapeutic protein. Under these conditions, circulating and resident immune cells produce lysosomal enzyme (i.e., IDUA) that can gain access to local and distant tissues via circulation in the plasma with M6P uptake and GAG clearance. Analytics from patients undergoing alloBMT have shown decreased urinary GAGs, improved organomegaly and stabilization of neurological disease [38]. However, residual disease can persist and whether that is due to the comparatively low levels of lysosomal enzyme provided by engrafted cells or from regimen-related toxicity is not fully defined [38]. Since 2002, total body irradiation-based conditioning has been replaced with busulfan/cyclophosphamide or, as of 2012, busulfan/fludarabine [39,40]. Therefore, as part of an evolving treatment regimen, current guidelines advocate early referral and intervention with the best possible HLA match with busulfan-conditioning regimens that facilitate maximal engraftment [38,41]. Continued follow-up will be required to determine whether physiological levels of enzyme or supranormal levels will result in the most complete responses. In support of the current human interventional guidelines, murine studies show that neonatal BMT with busulfan as a conditioning agent resulted in high engraftment and nearly absent skeletal disease [42], a common site of residual disease in MPS IH patients [38]. Corresponding adult animal studies showed that supraphysiological levels of enzyme achieved by lentiviral-mediated gene transfer of the IDUA gene to hematopoietic progenitors resulted in reversal of pathology in 6-week-old transplanted IDUA-/- [43] mice [44]. Comparative BMT controls did not exhibit as complete of a response, suggesting that a threshold level of enzyme can drive better outcomes in models where pathology is evident at the time of intervention [44].
The direct incorporation of therapeutic sequences into a patient’s own cell/genome represents a highly desirable strategy that will lower the risks inherent to unrelated donor cell therapies. At present, gene therapy and gene editing represent a path to the realization of this goal and are at the leading edge of next-generation therapies for the treatment of LSD.
Gene therapy classically refers to the provision of functional copies of a transgene under the control of exogenous regulatory elements and can encompass non-viral and viral integrating or episomal platforms. The cargo can be delivered directly to tissues or introduced into isolated cells for infusion back into the recipient. In vivo delivery of the SGSH gene via adeno-associated viral (AAV) vector is presently in Phase I/II clinical trials (NCT02716246) for MPS IIIA and relies on intravenous infusion of AAV serotype 9 particles that have tropism for multiple tissues including the CNS [45,46]. Delivery of AAV-10 particles via intracerebral injection has also been performed in MPS III patients (NCT01474343 and NCT02053064). While trials for stable gene transfer of repopulating hematopoietic progenitors have not yet reached the clinic for MPS, impetus for pursuing this course is provided by the treatment of the LSD metachromatic leukodystrophy by lentiviral transduction of CD34+ hematopoietic stem/progenitor cells (HSPC) [47]. This strategy halted the disease process and resulted in supranormal therapeutic gene/protein expression levels [47]. These studies highlight the therapeutic value of autologous avenues of investigation; however, one of the key attributes of AAV- episomal-based gene expression- may also be limiting for lifelong therapy due to the potential for loss of vector genomes by cellular division. The integrating properties of lentiviral vectors allow for sustained gene expression with transmittance of the transgene to progeny cells; however, the bias toward integrating into areas of transcriptional activity represents a significant safety risk [48–51]. As such, gene therapy approaches can be limiting and an ideal strategy would be to merge the attributes of AAV, limited ectopic integration and efficient delivery, with the permanence achieved with lentiviral-based approaches without the associated genotoxic risk. Programmable gene editing nucleases operate within this spectrum and are poised to make an impact in autologous MPS therapies.
Gene editing reagents are DNA binding proteins that allow for precision sequence modification facilitated by the induction of targeted double-stranded DNA breaks (DSB). These breaks stimulate DNA repair by error prone non-homologous endjoining or the more specific homologous recombination (HR) repair pathway [52]. HR is a DNA repair mechanism that utilizes homologous sequences as the repair template [53]. In the context of genome engineering, an artificial, exogenous donor can be provided in order to seamlessly incorporate user-defined sequences proximal to the DNA break. There are four main classes of gene editing proteins with differential architecture but common modes of action: meganucleases (MNs), zinc finger nucleases (ZFNs), transcription activator like effector nucleases (TALENs) and clustered regularly interspaced palindromic repeats (CRISPR)/Cas9. Each has a DNA binding component that can be programmed for a specific genomic target where it can then cleave one or both strands of DNA.
ZFNs are comprised of three to six finger subunits made of approximately 30 amino acids that each bind three nucleotides of DNA [54]. ZFNs are heterodimeric arrays that are tethered to the FokI nuclease domain that mediates the DSB upon target site dimerization [55]. Similar to ZFNs, TALENs are dimeric proteins that also rely on FokI-mediated endonuceolytic cleavage. Their DNA binding is directed by monomeric TAL proteins that each bind a single nucleotide of DNA and this interaction is dictated by a hypervariable two amino acid repeat region in each TAL subunit [56–58]. MNs are monomeric enzymes derived from bacterial homing endonucleases (HE) that can be repurposed for gene-specific targeting via a complicated engineering process constructed around a ‘central 4’ base pair recognition sequence that is variable between the individual classes of HE [59,60]. CRISPR/Cas9 is a bacterial adaptive immune system that is mobilized as a phage defense mechanism. Primary infection results in acquisition of phage sequences into the bacterial genome between palindromic repeats [61–63]. This sequence allows for the bacterium to differentiate self from non-self in order to cleave subsequent invading phage DNA without compromising the integrity of its own genome [64]. CRISPR/Cas9 has been repurposed for mammalian cell application and species variants derived from Streptococcus pyogenes [65,66] and Staphylococcus aureus [67] are the dominant candidates in use to date.
Gene editing reagents can be considered for the emergent and existing MPS patient populations and can be employed for ex vivo cellular modification or in vivo gene correction. Significant progress in ex vivo patient-derived HSPC modification has been made since the first efforts using ZFNs showed low rates of HDR (<1%) [68]. By optimizing HSPC ex vivo culture conditions with the addition of agents that promote HSPC expansion, homing and survival (StemRegenin1 and P16,16-dimethyl-prostaglandin E2) [69–71], Genovese et al. showed IL2RG gene modification by HR with secondary engraftment in immune-deficient mice [72]. Important for this was a sequential delivery of the donor fragment packaged in an integrase deficient lentivirus followed by ZFN mRNA [72]. Alternative donor delivery platforms have also been employed in an iterative manner with ZFN mRNA. Utilizing AAV serotype 6 (AAV-6) particles Wang and colleagues achieved rates of 17–19% at CCR5 and 26–43% at the AAVS1 loci in HSPC obtained from peripheral blood or fetal liver, respectively [73]. Importantly, this study showed that bulk isolation and treatment resulted in modification of the CD34+CD133+CD90+ sub-population that defines the primitive, highly engraftable hematopoietic precursors [73]. megaTAL nucleases (a hybrid molecule of a MN and TAL DNA binding arrays) have also facilitated high (~35%) rates of HR in HSPC at the CCR5 locus combined with AAV-6 donor delivery [74]. Recently, CRISPR/Cas9 has also been shown to mediate HR in HSPC that reached maximal rates with electroporation-based transfer of the Cas9 protein complexed with a gRNA targeting the β-globin (HBB) gene followed by incubation with a donor template encapsulated in AAV-6 viral particles [75]. Thus, the experimental conditions defined in disparate disorders for HSC correction will allow for rapid translation to LSD/MPS.
An important consideration will be the selection of gene modification events/cells in order to achieve maximal engraftment with cells expressing functional enzyme. The Genovese study shows a novel approach by fusing downstream exons that are spliced into upstream endogenous ones to reconstitute the full open reading frame [72]. As such, the introduction of a marker molecule allowing for selection could be employed in tandem with partial open reading frames to functionally correct a gene target (Figure 2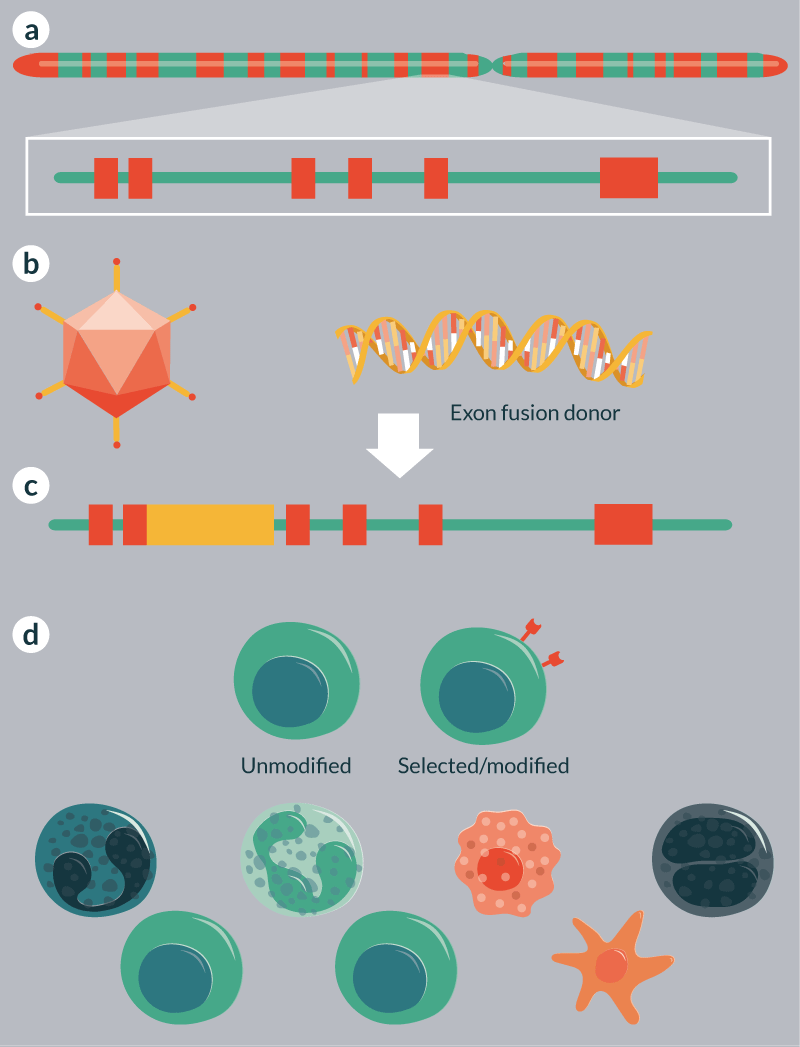
Gene therapy studies show that in vivo infusion of retroviral [78], lentiviral [79] and AAV [80] can also result in widespread gene expression and a concomitant absence of onset of disease. The innate tropism of viral vectors facilitates broad biodistribution making direct in vivo gene modification with programmable nucleases possible. Rather than circulating cells providing the enzyme, tissues would provide the enzyme locally with secreted enzyme that gains access to the circulation available for uptake from distant cells. Elegant studies using ZFNs [81,82] and CRISPR/Cas9 [83,84] with AAV delivery vehicles show in vivo gene targeting in murine models of hemophilia [81,82], Duchene muscular dystrophy (DMD) [83] and ornithine transcarbamylase deficiency (OTC) [84], respectively.
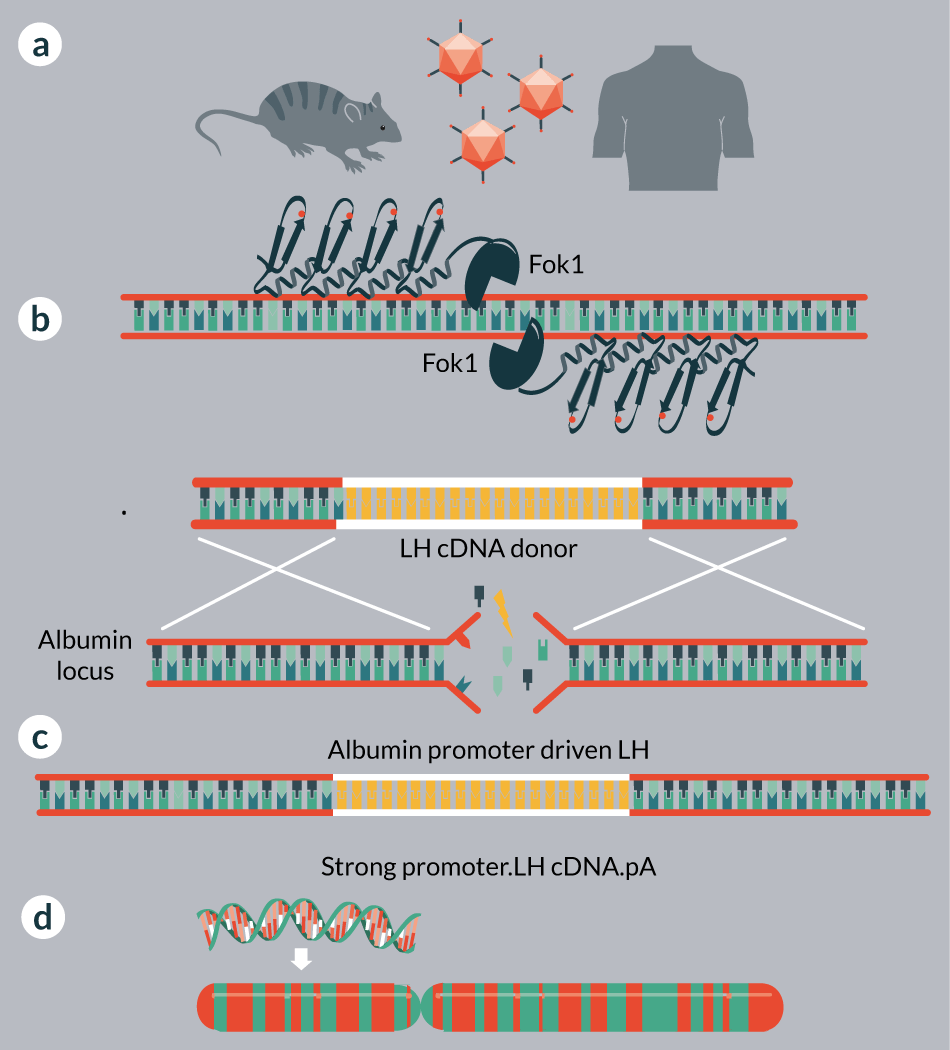
The DMD and OTC strategies target regions proximal to a disease causing alteration and gene correction occurs in situ such that the endogenous regulatory regions retain control of the gene. The DMD study relied on non-homologous endjoining for exon 23 deletion and in frame reconstitution of the remaining sequence [83]. The OTC targeting schema relied on HR-mediated repair [84]. Therefore, direct targeting of an MPS causing mutation could result in reversion to wild type status in vivo. The approach employed for hemophilia is a novel strategy relying on nuclease mediated insertion of a promoterless expression cassette into the albumin locus (Figure 3) [82]. By usurping the albumin promoter for enzyme production therapeutic levels of factor VIII protein (~40% of normal) have been achieved in murine models in vivo [82]. This study also showed that MPS lysosomal enzyme encoding genes (IDUA and IDS) could be employed as well and form the basis for clinical use in MPS I (NCT02702115) and MPS II (SB-913) patients. The experimental and clinical studies are designed to deliver the cargo on three AAV vectors (two each for the ZFN monomers and one for the donor), resulting in transgene expression from the strong albumin promoter in hepatocytes [81,82]. Thus, on a per cell basis supranormal levels of enzyme are produced that at the organismal level provide therapeutic levels of enzyme.
Both in vitro and ex vivo cellular modification have important considerations that will impact their employment: i) the capacity of a given reagent to be effectively delivered for therapy; and ii) the safety profile of the reagent. For mutation-specific targeting in isolated cells all of the nuclease reagents can be delivered as mRNA and a primary consideration becomes vectorability for in vivo delivery. TALENs are the largest gene-editing reagent and exceed the cargo capacity of AAV. Likewise, poor stability and titers have been reported for TALENs expressed from lentiviral vectors [85]. Both MNs and ZFNs are suitable for delivery in any size-restricted vector and the CRISPR/Cas9 derived from S. aureus is suitable for AAV packaging [67,86]. Further, Cas9 protein complexed with a gRNA has shown to be an efficient platform for in vitro delivery and modification [75]. Therefore, key considerations for achieving the desired cell modification event include: the targeting requirements and capability of a given reagent to be designed for that sequence; the associated time and cost of candidate engineering; and the manner in which the functional material will/can be delivered.
Gene editing reagents are programmed for a user-defined sequence of DNA; however, off-target effects for each class of nuclease have been observed due to the presence of partially homologous sequences in the genome [87–90]. As such, rigorous quality assurance and control to assess the on-target safety profile of gene editing proteins is an important clinical question and will factor in whether specific mutations are targeted individually or more broadly applicable platforms (e.g., safe harbor) are employed. At present, both approaches have attributes and drawbacks: mutation-specific targeting in the context of the broad mutational landscape of LSDs may represent a burden for development and deployment. However, the putative broad use/safe harbor reagents (e.g., albumin and AAVS1) have demonstrated off-target effects [81,91] and the notion of a true safe harbor is subject to debate [92]. This may be concerning because the donor sequences that have regulatory elements may have an amplified risk of gene disruption due to activation or inactivation [49,50]. For mutation targeting with donor sequences that have no regulatory elements the ramification of off-target incorporation is essentially limited to random gene disruption and inactivation.
The optimization of reagents with greater specificity and capability will co-evolve with the identification of robust deployment strategies to achieve the most effective enzyme delivery platform for autologous therapies. The expansive functionality of gene editing reagents, in particular the CRISPR/Cas9 system, also represents a tool for the basic, translational and clinical aspects of comprehensive MPS therapy development. Ongoing basic biological investigations to uncover the underpinnings of the GAG-induced secondary effects have shown the promise of mechanism-based agents for targeting refractory and residual disease [93,94]. As such, in addition to primary gene correction by HR, the ability to knockout or modulate the expression levels of genes will allow for more accurate modeling and discovery of genes that impact or can be targeted for enhanced therapeutic outcomes. The value of isogenic cell line generation by ZFNs has been demonstrated in Parkinson’s disease [95] and genome wide knockout [96] and transcriptional activation [97] screens have identified novel candidates for therapies. Therefore, the flexibility and versatility of genome editing reagents can be brought to bear in a similar fashion for LSDs. By deploying these reagents in the optimal therapeutic window gene editing nucleases have tremendous potential for the treatment of MPS and will serve as a platform for rapid translation for many other disorders.
Financial & competing interests disclosure
The authors have no relevant financial involvement with an organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock options or ownership, expert testimony, grants or patents received or pending, or royalties. No writing assistance was utilized in the production of this manuscript.
References
1. de Duve C. The lysosome turns fifty. Nat. Cell Biol. 2005; 7: 847–9.
CrossRef
2. De Duve C, Pressman BC, Gianetto R et al. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 1955; 60: 604–17.
CrossRef
3. Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu. Rev. Med. 2015; 66: 471–86.
CrossRef
4. Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nat. Rev. Mol. Cell Biol. 2004; 5: 554–65.
CrossRef
5. Meikle PJ, Hopwood JJ, Clague AE et al. Prevalence of lysosomal storage disorders. JAMA 1999; 281: 249–54.
CrossRef
6. Al-Jasmi FA, Tawfig N, Berniah A et al. Prevalence and Novel Mutations of Lysosomal Storage Disorders in United Arab Emirates: LSD in UAE. JIMD Rep. 2013; 10: 1–9.
CrossRef
7. Poupetova H, Ledvinova J, Berna L et al. The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. J. Inherit. Metab. Dis. 2010; 33: 387–96.
CrossRef
8. Moore D, Connock MJ, Wraith E et al. The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK. Orphanet J. Rare Dis. 2008; 3: 24.
CrossRef
9. Jurecka A, Lugowska A, Golda A et al. Prevalence rates of mucopolysaccharidoses in Poland. J. Appl. Genet. 2015; 56: 205–10.
CrossRef
10. Sasisekharan R, Raman R, Prabhakar V. Glycomics approach to structure-function relationships of glycosaminoglycans. Annu. Rev. Biomed. Eng. 2006; 8: 181–231.
CrossRef
11. Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 2007; 446: 1030–7.
CrossRef
12. Neufeld EF MJ. The mucopolysaccharidoses. In: The Metabolic and Molecular Bases of Inherited Disease. 2001; 3421–52, McGraw-Hill, NY, USA.
13. Coutinho MF, Lacerda L, Alves S. Glycosaminoglycan storage disorders: a review. Biochem. Res. Int. 2012; 2012: 471325.
14. Desnick RJ, Thorpe SR, Fiddler MB. Toward enzyme therapy for lysosomal storage diseases. Physiol. Rev. 1976; 56: 57–99.
15. Walkley SU. Pathogenic mechanisms in lysosomal disease: a reappraisal of the role of the lysosome. Acta Paediatr. Suppl. 2007; 96: 26–32.
CrossRef
16. Lieberman AP, Puertollano R, Raben N et al. Autophagy in lysosomal storage disorders. Autophagy 2012; 8: 719–30.
CrossRef
17. Shimada Y, Klionsky DJ. Autophagy contributes to lysosomal storage disorders. Autophagy 2012; 8: 715–6.
CrossRef
18. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140: 313–26.
CrossRef
19. Platt FM, Boland B, van der Spoel AC. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J. Cell. Biol. 2012; 199: 723–34.
CrossRef
20. Schultz ML, Tecedor L, Chang M et al. Clarifying lysosomal storage diseases. Trends Neurosci. 2011; 34: 401–10.
CrossRef
21. Vitry S, Bruyere J, Hocquemiller M et al. Storage vesicles in neurons are related to Golgi complex alterations in mucopolysaccharidosis IIIB. Am. J. Pathol. 2010; 177: 2984–99.
CrossRef
22. Pereira VG, Gazarini ML, Rodrigues LC et al. Evidence of lysosomal membrane permeabilization in mucopolysaccharidosis type I: rupture of calcium and proton homeostasis. J. Cell. Physiol. 2010; 223: 335–42.
CrossRef
23. Villani GR, Chierchia A, Di Napoli D et al. Unfolded protein response is not activated in the mucopolysaccharidoses but protein disulfide isomerase 5 is deregulated. J. Inherit. Metab. Diss. 2012; 35: 479–93.
24. Higa A, Taouji S, Lhomond S et al. Endoplasmic reticulum stress-activated transcription factor ATF6alpha requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol. Cell. Biol. 2014; 34: 1839–49.
CrossRef
25. Hayano T, Kikuchi M. Molecular cloning of the cDNA encoding a novel protein disulfide isomerase-related protein (PDIR). FEBS Lett. 1995; 372: 210–4.
CrossRef
26. Martins C, Hulkova H, Dridi L et al. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain 2015; 138: 336–55.
CrossRef
27. Villani GR, Di Domenico C, Musella A et al. Mucopolysaccharidosis IIIB: oxidative damage and cytotoxic cell involvement in the neuronal pathogenesis. Brain Res. 2009; 1279: 99–108.
CrossRef
28. Donida B, Marchetti DP, Biancini GB et al. Oxidative stress and inflammation in mucopolysaccharidosis type IVA patients treated with enzyme replacement therapy. Biochim. Biophys. Acta 2015; 1852: 1012–9.
29. Haq E, Contreras MA, Giri S et al. Dysfunction of peroxisomes in twitcher mice brain: a possible mechanism of psychosine-induced disease. Biochem. Biophys. Res. Commun. 2006; 343: 229–38.
CrossRef
30. Schedin S, Sindelar PJ, Pentchev P et al. Peroxisomal impairment in Niemann-Pick type C disease. J. Biol. Chem. 1997; 272: 6245–51.
CrossRef
31. Tatsumi K, Saito M, Lin B et al. Enhanced expression of a-series gangliosides in fibroblasts of patients with peroxisome biogenesis disorders. Biochim. Biophys. Acta 2001; 1535: 285–93.
CrossRef
32. McGlynn R, Dobrenis K, Walkley SU. Differential subcellular localization of cholesterol, gangliosides, and glycosaminoglycans in murine models of mucopolysaccharide storage disorders. J. Comp. Neurol. 2004; 480: 415–26.
CrossRef
33. Perera RM, Zoncu R. The Lysosome as a Regulatory Hub. Annu. Rev. Cell. Dev. Biol. 2016; 32: 223–53.
CrossRef
34. Lim CY, Zoncu R. The lysosome as a command-and-control center for cellular metabolism. J. Cell. Biol. 2016; 214: 653–64.
CrossRef
35. Fratantoni JC, Hall CW, Neufeld EF. The defect in Hurler and Hunter syndromes. II. Deficiency of specific factors involved in mucopolysaccharide degradation. Proc. Natl Acad. Sci. USA 1969; 64: 360–6.
CrossRef
36. Fratantoni JC, Hall CW, Neufeld EF. Hurler and Hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science 1968; 162: 570–2.
CrossRef
37. Hobbs JR, Hugh-Jones K, Barrett AJ et al. Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet 1981; 2: 709–12.
CrossRef
38. Aldenhoven M, Wynn RF, Orchard PJ et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood 2015; 125: 2164–72.
CrossRef
39. Aldenhoven M, Jones SA, Bonney D et al. Hematopoietic cell transplantation for mucopolysaccharidosis patients is safe and effective: results after implementation of international guidelines. Biol. Blood Marrow Transplant. 2015; 21: 1106–9.
CrossRef
40. Bartelink IH, van Reij EM, Gerhardt CE et al. Fludarabine and exposure-targeted busulfan compares favorably with busulfan/cyclophosphamide-based regimens in pediatric hematopoietic cell transplantation: maintaining efficacy with less toxicity. Biol. Blood Marrow Transplant. 2014; 20: 345–53.
CrossRef
41. Boelens JJ, Aldenhoven M, Purtill D et al. Outcomes of transplantation using various hematopoietic cell sources in children with Hurler syndrome after myeloablative conditioning. Blood 2013; 121: 3981–7.
CrossRef
42. Pievani A, Azario I, Antolini L et al. Neonatal bone marrow transplantation prevents bone pathology in a mouse model of mucopolysaccharidosis type I. Blood 2015; 125: 1662–71.
CrossRef
43. Clarke LA, Russell CS, Pownall S et al. Murine mucopolysaccharidosis type I: targeted disruption of the murine alpha-L-iduronidase gene. Hum. Mol. Gen. 1997; 6: 503–11.
CrossRef
44. Visigalli I, Delai S, Politi LS et al. Gene therapy augments the efficacy of hematopoietic cell transplantation and fully corrects mucopolysaccharidosis type I phenotype in the mouse model. Blood 2010; 116: 5130–9.
CrossRef
45. Schuster DJ, Dykstra JA, Riedl MS et al. Biodistribution of adeno-associated virus serotype 9 (AAV9) vector after intrathecal and intravenous delivery in mouse. Front. Neuroanat. 2014; 8: 42.
CrossRef
46. Pacak CA, Sakai Y, Thattaliyath BD et al. Tissue specific promoters improve specificity of AAV9 mediated transgene expression following intra-vascular gene delivery in neonatal mice. Genet. Vaccines Ther. 2008; 6: 13.
CrossRef
47. Biffi A, Montini E, Lorioli L et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013; 341: 1233158.
CrossRef
48. Cattoglio C, Facchini G, Sartori D et al. Hot spots of retroviral integration in human CD34+ hematopoietic cells. Blood 2007; 110: 1770–8.
CrossRef
49. Hacein-Bey-Abina S, Garrigue A, Wang GP et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008; 118: 3132–42.
CrossRef
50. Hacein-Bey-Abina S, Von Kalle C, Schmidt M et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003; 302: 415–9.
CrossRef
51. Cavazzana-Calvo M, Payen E, Negre O et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 2010; 467: 318–22.
CrossRef
52. Elliott B, Richardson C, Winderbaum J et al. Gene conversion tracts from double-strand break repair in mammalian cells. Mol. Cell. Biol. 1998; 18: 93–101.
CrossRef
53. Guirouilh-Barbat J, Lambert S, Bertrand P et al. Is homologous recombination really an error-free process? Front. Genet. 2014; 5: 175.
CrossRef
54. Beerli RR, Barbas CF, 3rd. Engineering polydactyl zinc-finger transcription factors. Nature Biotechnol. 2002; 20: 135–41.
CrossRef
55. Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl Acad. Sci. USA 1996; 93: 1156–60.
CrossRef
56. Boch J, Scholze H, Schornack S et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009; 326: 1509–12.
CrossRef
57. Bogdanove AJ, Voytas DF. TAL effectors: customizable proteins for DNA targeting. Science 2011; 333: 1843–6.
CrossRef
58. Streubel J, Blucher C, Landgraf A et al. TAL effector RVD specificities and efficiencies. Nature Biotechnol. 2012; 30: 593–5.
CrossRef
59. Baxter S, Lambert AR, Kuhar R et al. Engineering domain fusion chimeras from I-OnuI family LAGLIDADG homing endonucleases. Nucleic Acids Res. 40: 7985–8000.
CrossRef
60. Stoddard BL. Homing endonucleases: from microbial genetic invaders to reagents for targeted DNA modification. Structure 19: 7–15.
CrossRef
61. Bolotin A, Quinquis B, Sorokin A et al. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 2005; 151: 2551–61.
CrossRef
62. Mojica FJ, Diez-Villasenor C, Garcia-Martinez J et al. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005; 60: 174–82.
CrossRef
63. Pourcel C, Salvignol G, Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 2005; 151: 653–63.
CrossRef
64. Marraffini LA, Sontheimer EJ. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 2010; 463: 568–71.
CrossRef
65. Mali P, Yang L, Esvelt KM et al. RNA-guided human genome engineering via Cas9. Science 2013; 339: 823–6.
CrossRef
66. Cong L, Ran FA, Cox D et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013; 339: 819–23.
CrossRef
67. Ran FA, Cong L, Yan WX et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015; 520: 186–91.
CrossRef
68. Lombardo A, Genovese P, Beausejour CM et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nature Biotechnol. 2007; 25: 1298–306.
CrossRef
69. Goessling W, Allen RS, Guan X et al. Prostaglandin E2 enhances human cord blood stem cell xenotransplants and shows long-term safety in preclinical nonhuman primate transplant models. Cell Stem Cell 2011; 8: 445–58.
CrossRef
70. North TE, Goessling W, Walkley CR et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature 2007; 447: 1007–11.
CrossRef
71. Boitano AE, Wang J, Romeo R et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science 2010; 329: 1345–8.
CrossRef
72. Genovese P, Schiroli G, Escobar G et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature 2014; 510: 235–40.
CrossRef
73. Wang J, Exline CM, DeClercq JJ et al. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat. Biotechnol. 2015; 33: 1256–63.
CrossRef
74. Sather BD, Romano Ibarra GS, Sommer K et al. Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci. Transl. Med. 2015; 7: 307ra156.
CrossRef
75. Dever DP, Bak RO, Reinisch A et al. CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature 2016; 539: 384–9.
CrossRef
76. Hockemeyer D, Soldner F, Beard C et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009; 27: 851–7.
CrossRef
77. Rio P, Banos R, Lombardo A et al. Targeted gene therapy and cell reprogramming in Fanconi anemia. EMBO Mol. Med. 2014; 6: 835–48.
CrossRef
78. Liu Y, Xu L, Hennig AK et al. Liver-directed neonatal gene therapy prevents cardiac, bone, ear, and eye disease in mucopolysaccharidosis I mice. Mol. Ther. 2005; 11: 35–47.
CrossRef
79. Derrick-Roberts AL, Pyragius CE, Kaidonis XM et al. Lentiviral-mediated gene therapy results in sustained expression of beta-glucuronidase for up to 12 months in the gus(mps/mps) and up to 18 months in the gus(tm(L175F)Sly) mouse models of mucopolysaccharidosis type VII. Hum. Gene Ther. 2014; 25: 798–810.
CrossRef
80. Daly TM, Ohlemiller KK, Roberts MS et al. Prevention of systemic clinical disease in MPS VII mice following AAV-mediated neonatal gene transfer. Gene Ther. 2001; 8: 1291–8.
CrossRef
81. Li H, Haurigot V, Doyon Y et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature 2011; 475: 217–21.
CrossRef
82. Sharma R, Anguela XM, Doyon Y et al. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood 2015; 126: 1777–84.
CrossRef
83. Nelson CE, Hakim CH, Ousterout DG et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016; 351: 403–7.
CrossRef
84. Yang Y, Wang L, Bell P et al. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nature Biotechnol. 2016; 34: 334–8.
CrossRef
85. Holkers M, Maggio I, Liu J et al. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013; 41: e63.
CrossRef
86. Friedland AE, Baral R, Singhal P et al. Characterization of Staphylococcus aureus Cas9: a smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol. 2015; 16: 257.
CrossRef
87. Guilinger JP, Pattanayak V, Reyon D et al. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat. Methods 2014; 11: 429–35.
CrossRef
88. Osborn MJ, Webber BR, Knipping F et al. Evaluation of TCR Gene Editing achieved by TALENs, CRISPR/Cas9 and megaTAL nucleases. Mol. Ther. 2015; 24: 570–81.
CrossRef
89. Gabriel R, Lombardo A, Arens A et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotechnol. 29: 816–23.
CrossRef
90. Cho SW, Kim S, Kim Y et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Gen. Res. 2014; 24: 132–41.
CrossRef
91. Tan EP, Li Y, Velasco-Herrera Mdel C et al. Off-target assessment of CRISPR-Cas9 guiding RNAs in human iPS and mouse ES cells. Genesis 2015; 53: 225–36.
CrossRef
92. Sadelain M, Papapetrou EP, Bushman FD. Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer 2012; 12: 51–58.
93. Hawkins-Salsbury JA, Shea L, Jiang X et al. Mechanism-based combination treatment dramatically increases therapeutic efficacy in murine globoid cell leukodystrophy. J. Neurosci. 2015; 35: 6495–505.
CrossRef
94. Osborn MJ, Webber BR, McElmurry RT et al. Angiotensin receptor blockade mediated amelioration of mucopolysaccharidosis type I cardiac and craniofacial pathology. J. Inherit. Metab. Dis. 2016 (Epub ahead of print).
CrossRef
95. Soldner F, Laganiere J, Cheng AW et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 2011; 146: 318–31.
CrossRef
96. Shalem O, Sanjana NE, Hartenian E et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014; 343: 84–7.
CrossRef
97. Konermann S, Brigham MD, Trevino AE et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015; 517: 583–8.
CrossRef
98. Ma X, Tittiger M, Knutsen RH et al. Upregulation of elastase proteins results in aortic dilatation in mucopolysaccharidosis I mice. Mol. Genet. Metab. 2008; 94: 298–304.
CrossRef
99. Baldo G, Wu S, Howe RA et al. Pathogenesis of aortic dilatation in mucopolysaccharidosis VII mice may involve complement activation. Mol. Genet. Metab. 2011; 104: 608–19.
CrossRef
100. Braunlin EA, Harmatz PR, Scarpa M et al. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J. Inherit. Metab. Dis. 2011; 34: 1183–97.
CrossRef
Affiliations
Mark J Osborn1–5, Dok Hyun Yoon5–7, Beom Hee Lee8, Chong Jai Kim5,7 & Jakub Tolar1–5
1 Department of Pediatrics, University of Minnesota Medical School, Division of Blood and Marrow Transplantation, Minneapolis, MN, USA
2 Masonic Cancer Center, University of Minnesota, Minneapolis, MN, USA
3 Stem Cell Institute, University of Minnesota, Minneapolis, MN, USA
4 Center for Genome Engineering, University of Minnesota, Minneapolis, MN, USA
5 Asan-Minnesota Institute for Innovating Transplantation, Seoul, Republic of Korea
6 Department of Oncology, University of Ulsan College of Medicine, Asan Medical Center, Seoul, Republic of Korea
7 Asan Institute for Life Sciences, Seoul, Republic of Korea
8 Medical Genetics Center, Asan Medical Center Children’s Hospital, University of Ulsan College of Medicine, Seoul, Republic of Korea

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.