AAV vector production: state of the art developments and remaining challenges
Cell Gene Therapy Insights 2016; 2(5), 521-551.
10.18609/cgti.2016.067
In 2012, an adeno-associated virus (AAV) vector reached the market following its approval by the European Medicines Agency for the treatment of patients with lipoprotein lipase deficiency. This marketing authorisation represented an important step for the gene therapy field, moving from exploratory towards routine clinical use. However, a number of challenges remain in the production and manufacture of these therapies. In principle, four different production platforms (HEK293[T] transfection, stable packaging/producer cell lines, herpes simplex virus system and baculovirus system) have been developed and for some, scale-up to a 2000 L scale has already been performed. Despite these achievements and the well-advanced technological developments in manufacturing technology, further improvements in AAV technology are required to increase vector titers, and improve vector quality and potency. This article provides a review of the four vector production platforms, recent improvements and developments as well as perspectives on future requirements.
The in vivo use of AAV vectors for the treatment of rare diseases has come of age, in particular, with the successful outcomes of several clinical trials, including the treatment of hemophilia B [1–4] and Leber’s congenital amaurosis type 2 [5–9], and obviously with the marketing authorization of uniQure’s Glybera® (an AAV1-based gene therapy product for the treatment of familial lipoprotein lipase deficiency) in the EU in Autumn 2012 [10]. There are many additional clinical applications in development that utilize AAV vectors for the treatment of various rare and acquired diseases and up to February 2016, 162 clinical trials using AAV vectors have been performed, which equates to 6.7% of all clinical gene therapy trials performed at that point (http://www.abedia.com/wiley/vectors.php). These promising developments and achievements were only possible because advances in bioprocessing and analytical development provided the necessary tools for the manufacture and quality control of clinical-grade AAV vectors.
Most of the AAV vector production to date has been performed at a small–medium scale using transfection-based production methods. These methods allow for the production of AAV vector at sufficient quantities for the treatment of diseases such as those of the retina, or for Phase I clinical trials, as in the case of the treatment of hemophilia B. In more advanced clinical trials or when moving to more routine use of AAV vectors for marketing purposes, larger vector amounts are required, which cannot be produced (or can only be produced with difficulty) using the traditional transfection method.
In order to design an adapted and scalable production method, it is important to know the vector quantity required for the clinical trial, which obviously depends on the dosing and the number of subjects to be treated. This is explained in more detail with the following two examples:
- A Hemophilia B trial carried out by Nathwani et al.[1,2] used an scAAV8 vector produced via bi-transfection of HEK293T cells grown in CS10 (10-stack CellSTACKS®, SigmaAldrich) [11]. In order to produce the required vector quantity for the treatment of the first six patients (two patients per cohort; in total, three different doses were assessed [2 x 1011 vg/kg patient weight; 6 x 1011 vg/kg; and 2 x 1012 vg/kg]) [1] and the subsequent four patients treated with the high dose [2], two production campaigns of a total duration of 18 months consisting of 432 independent 10-stack culture devices were performed [11].
- A preclinical study performed in GRMD (the dog model of Duchenne Muscular Dystrophy [DMD]) using an U7-based exon skipping approach vectorized by an ssAAV8 vector and produced with the insect cell/baculovirus system [12] indicated that an efficient dose should be beyond 5 x 1013 vg/kg (= high dose in that study). At this dose, approximately 40% of the muscle fibers became dystrophin positive after locoregional administration [13]. If this vector amount should be produced with the traditional transfection system for a Phase 1 clinical trial with three cohorts of three patients (with dose progression, highest dose: 5 x 1013 vg/kg), 1250–2500 10-stack devices would be needed.
Both examples clearly demonstrate that either for advanced clinical trials as for hemophilia B or for the treatment of muscular tissues representing approximately 50% of the whole body tissue such as in the case of neuromuscular diseases (e.g., DMD), huge amounts of AAV vectors will be needed, which have to be produced with adapted methods. It is obvious that the production of the scAAV8 vector as used in the hemophilia B study of Nathwani et al. [1-3] was limited with respect to the production system, and producing larger AAV quantities with that method cannot be readily envisaged. With respect to the preclinical trials assessing the exon skipping approach for the treatment of DMD [13], the choice of the insect cell baculovirus system was adapted because this production system is scalable.
On the other hand, both studies provided insights into the vector quantities required for the transduction of liver or muscle tissues and it is obvious that for the treatment of muscular dystrophies, huge amounts of AAV vectors are needed, which can only be produced by optimized scalable production systems. However, another perhaps less obvious issue is that generally the efficiency and potency of AAV vector lots has to be increased, which will lead to the reduction in the effective doses. This review presents an update on the available large-scale production and purification methods, their advantages and limitations, as well as challenges to overcome.
Biological systems for the production of AAV vectors
AAV is a single-stranded DNA virus of the Dependovirus genus, part of the Parvovirus family. The name of the genus derives from the fact that AAV replication depends on the presence of a helper virus, such as adenovirus or herpes simplex virus (HSV), signifying that for vector production the required helper functions from one of these virus have to be provided. Since the development of the baculovirus expression system for AAV production (see below), it has become apparent that the baculovirus can also provide the required helper function. Another important point to note is that the overall packaging capacity of AAV vectors is rather small (approximately 4.7 Kb including the two inverted terminal repeats [ITRs]). Its overall organization is presented in Figure 1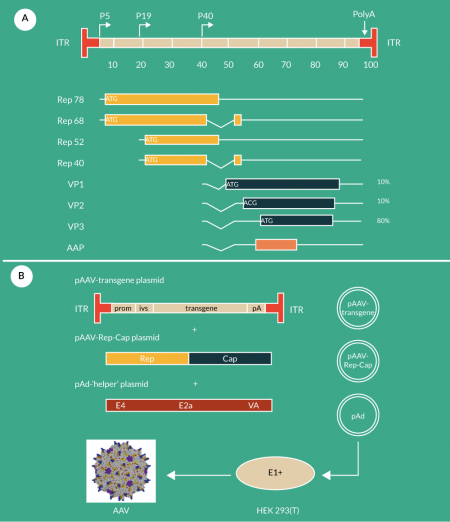
The first production system developed was based on the transfection of HEK293(T) cells with two or three plasmids. Although this system is very versatile and allows for the simple modification of the capsid and/or recombinant vector sequence, its main drawback is the limited scalability; therefore other systems have been developed to address this limitation: the use of stable packaging and producer cell lines; the recombinant HSV Type 1 expression system; or the use of the baculovirus/insect cell system. These systems will be discussed in more detail in the following paragraphs.
Traditional methods for the production of AAV vectors: transfection of HEK293(T) cells
Use of adherently growing cells
The traditional production methods for recombinant AAV (rAAV) are based on transient Calcium-phosphate based transfection of HEK293(T) cells. This typical laboratory approach is based on the delivery of the AAV rep and cap functions, of the adenoviral helper functions E2a, E4orf6 and VA [14,15] and of the recombinant rAAV transgene construct (in which the two ITRs flank the recombinant vector construct of a maximum size of 4.5 kB) to the HEK293(T) cells (Figure 1B). These cells constitutively express the adenoviral E1a/E1b functions, which are also involved in the synthesis of rAAV vectors.
While the original system is based on the use of three plasmids (pAAV-rep-cap, pAd-helper and pAAV-transgene Figure 1B), a streamlined dual system was developed by combining the two helper plasmids into one plasmid [17]. The main advantage is that the cells only have to be transfected with two different plasmids. This system requiring only a double hit cell transfection results in up to ten-fold higher titers than those obtained with the conventional protocols [17].
Traditionally, transfection-based production is performed in 15-cm cell culture plates, which allow the production of 1011 vg/plate. Since adherently growing cells are used, an increase in vector production is performed by an increase in the cell culture surface, which in practice is performed via the addition of parallel culture plates (a scale-out approach).
Further increases in production scale can be performed via the use of larger culture devices including CellFactory™ (ThermoFisher) or CellSTACKs® as used by Allay et al. [11] for the production of scAAV8 vectors for the hemophilia B study, or roller bottles as used for the production of ssAAV2 in the Leber’s congenital amaurosis type 2 trials [18]. In the example of the roller bottle process, a sub-batch of 100 roller bottles consisted of 0.5–1 x 1015 vg (purified vector) signifying that cell-specific vector production was approximately 2 x 105 vg/c [18]. Similar specific vector production rates have been reported for a CellSTACK process generating up to 1015 vg/lot of six stacks [19].
As already mentioned, this production method allows for the generation of vector quantities sufficient for a Phase I clinical trial as in the case of the scAAV8-based clinical trial for the treatment of hemophilia B for which 432 parallel CellSTACKs had to be used; however, this example also demonstrates the limitation of this approach when envisaging such a gene therapy approach for late-stage clinical trials or routine clinical use.
A recent development in vector manufacturing is the use of fixed bed reactor systems (iCELLis®, PALL – disposable system) for the production of viruses and viral vectors and in this context the production of AAV vectors via a transfection-based method can be achieved, although the application of a fixed bed reactor system is much simpler when stable producer cell lines are used (e.g., as with the production of retroviral vectors [20,21]). Nevertheless, it was demonstrated that HEK293T cells grown in a 2-cm fixed bed (iCELLis® Nano, 0.53 m2) could be transfected homogenously and generated in average 3.6 x 1014 ± 7.1 x 1013 viral particles, which was approximately a quarter of the quantity generated with 15-cm culture dishes (at equal surface) [22]. In comparison, the iCELLis® Nano system produced two-times lower rAAV levels than a 10-stack CellFactory™ system: 2.2 x 1010 and 4.7 x 1010 vg/cm2, respectively [23]. An iCELLis Nano unit thus produced 1.14 x 1014 vg per run (without optimization). Our own studies confirmed the utility of the iCELLis® system for AAV vector production and also showed that the 10-cm bed system could be relatively homogenously transfected [24]. The advantage of the iCELLis® system is obviously its scalability up to the iCELLis® 500 system, which provides a surface of 500 m2 at the high compaction version, meaning that for adherently growing cells, the iCELLis® system represents a real breakthrough with respect to scale up. Further advantages are the control of the culture with respect to pH, pO2, agitation and aeration, as well as the fact that much less operator involvement is required when compared to the use of roller bottles or CellFactory™ systems. However, several issues are related to scale-up of the iCELLis 500 system that are yet to be resolved. In the case of obligatory adherent cells, cell amplification can only be performed using adapted systems such as roller bottle or CellFactories or more sophisticated systems such as Pall’s Xpansion® Bioreactor system. In the case of cells lines that can also grow in suspension, cell expansion is simply performed by suspension culture using wave type reactors, for instance. In contrast to Emmerling et al. [23] we have used a serum-free culture medium (a modified F17 medium for cell growth and DMEM for virus production in order to boost AAV vector production), which renders the process adapted for the production of clinical material [24]. Furthermore, the harvest of AAV vectors can be simplified by using a detergent containing harvest buffer for cell lysis [12], meaning that under these conditions, the process is easily scalable for routine manufacturing of AAV vectors for clinical use.
Use of suspension culture
Another and more flexible approach to the scale-up of transfection-based production processes is the use of suspension culture for which scale-up is much easier to perform than for surface adherent cell cultures. However, in this context, the use of the classical Ca-phosphate transfection protocol had to be replaced because of the huge impact on transfection efficiency resulting from environmental variations in pH, Ca2+ and phosphate concentrations. Furthermore, Ca-phosphate is cytotoxic whereby serum or albumin must therefore be present, and second, a complete medium exchange is required prior to transfection [25]. Since this is not really feasible at a large suspension culture process, other transfection agents such as polyethylenimine (PEI) or cationic lipids are used that, in addition, provide much more reproducible results because they are independent of environmental/culture conditions.
Durocher et al. showed that AAV2 vectors could be produced in suspension cultures of HEK293E via triple transfection using PEI [26] and some years later the same group demonstrated that using the same process, the production of AAV serotypes 1–9 was possible, yielding product levels of 1013 vg/L [27]. These production levels could be confirmed at a 3 L reactor scale. Further scale-up of a suspension process protocol using 10- and 20-L WAVE reactors (GE Healthcare) was performed by Grieger et al. leading to vector yields in the range of 6.7 x 1012 to 3.5 x 1013 vg/L, depending on the AAV serotype and vector construct [28]. Total yields of up to 5.5 x 1014 vg could be achieved for 20 L cultures.
Although published data only represent production scales of up to 20 L [28], it is evident that scales as large as 200 L (or even larger) could be possible, allowing ten-times higher vector yields. However, it should be kept in mind that the costs of the GMP-grade plasmids (2 or 3) are much higher, meaning that they represent the most important production cost factor. A further drawback is the fact that transfection-based production methods and their yields are impacted to a larger extent by environmental factors than other production methods (see below).
Further developments & optimization
Although transfection-based large-scale production methods have been developed, further improvements are still possible and necessary in order to increase vector yields, vector quality (in particular, the full-to-empty-particle titer) and vector potency. These improvements include the choice of the cell line/cell clone, the plasmid construct, the choice of the medium and culture conditions.
Cells
Although the traceability of the HEK293(T) cells is relatively limited and incomplete [29], the routine use of this cell line for GMP production of different viral vectors is well established because of their advantageous growth behavior, transfectability and good capacity for vector production. Nevertheless, improvements are possible, for instance via the selection of better producing cell clones as carried out by Grieger et al., who selected a HEK293 clone capable of growing in suspension and characterized by high transfection efficiency and AAV vector production (>105 vg/c, >1014 vg/L) [28]. Although it can be inferred that HEK293T cells should be the more efficient cell line because of their superior proliferation capacity, their greater transfectability and higher vector production rate compared with HEK293 cells [29], the presence of the SV40 T-antigen might present an increased safety risk and therefore it might be judicious to favor the use of the HEK293 cells for AAV vector production.
Plasmids/plasmid constructs
Today, the two or three plasmid systems most commonly used are produced using traditional bacterial production systems [30]. Plasmids contain backbone sequences, which contain motifs, which are recognized by the cell-autonomous immune system and are therefore prone to induce gene silencing and inflammatory responses [31,32]. Furthermore, the sequence of antibiotic resistance genes might be encapsidated into the AAV vector particles and thus might be transferred to the target tissue [33]. The frequency of the presence of AAV particles containing a rAAV vector coding for the resistance gene, can be 2.9 and 26.1%, respectively, for single-strand AAV (ssAAV) and self-complementary AAV (scAAV) vector preparation [34]. In order to avoid the transfer of plasmid-derived sequences into the target tissue, Schnödt et al. have adapted Plasmid Factory’s DNA minicircle technology to the generation of the plasmids used for the AAV production using a two plasmid transfection-based approach [34]. The evaluation of this plasmid system could show that the total particle yield was comparable to the classical plasmid system but the vector preparations contained higher numbers of transgene sequence containing particles. Furthermore, the use of the minicircle technology resulted in vector preparations with superior transduction efficiency, in particular, in the case of the production of scAAV vectors. With respect to the encapsidation of residual plasmid sequences, only 1.3% of the vector capsids still contained the short residual prokaryotic non-coding SCAR sequence (213 bp), which contains one recombination sequence and a tag for affinity purification [35,36]. Ideally, this sequence should also be removed in order to generate AAV vector preparations devoid of particles with foreign DNA sequences derived from the plasmids.
On the other hand, the helper plasmids (as used in a double or triple transfection approach) were also improved [23]. With regards to the adenoviral helper construct (pUC AdV), unnecessary sequences were removed, leading to a size of 10236 bp instead of 15263 bp. The rep/cap AAV helper plasmid was split into two separate plasmids with the advantage of reducing the risk of the generation of rcAAV particles (split packaging approach). Furthermore, these plasmids have been optimized by inactivating dispensable promoters and deleting potential start codons in the cap open reading frames. This led to a rep plasmid (pUCrepoptΔrep78/Δcap) for the expression of rep68 and rep52/40 proteins via separate sequences (in both sequences, the p40 was inactivated) and a cap plasmid (pUCcapoptp5p19p40cap) for the expression of the VP proteins. In this construct, the p5 and p19 promoters had been inactivated. Side-by-side comparison of this improved plasmid system consisting of four plasmids (the rAAV vector plasmid combined with the optimized rep/cap split-packaging plasmids and the helper adenoviral plasmid) with the widely used two plasmid system of the pDG family [37–39] showed that the optimized plasmid system led to a 12-fold increase in vector yield (2.7 x 105 vg/c) when using HEK293T cells grown adherently in serum-containing medium; however, under serum-free suspension conditions, the viral vector yield was in the range of 2 x 104 vg/c, probably due to the insufficient composition of the medium or lack of an essential nutrient [40].
Although this might be the case for suspension processes where we know that the cells normally grown in an attached mode are more sensitive to nutrient limitations, several authors have reported that for transfection-based processes using adherently grown HEK293(T) cells, the switch from a serum-containing medium to a simple DMEM 24 h after transfection leads to elevated release of AAV vectors into the supernatant: AAV2/5 [41,42], AAV2/8 [43,44], AAV2/1, AAV2/7 and AAV2/9 [44]. This release could be prolonged for several days when regular medium harvests were performed [42]. It should be indicated here that for many AAV serotypes the release of vector particles into the culture supernatant is also observed when serum-containing media are used (AAV1, 7, 8, 9) [41]. From a functionality perspective, it could be demonstrated that the behavior of the supernatant and the cell-derived AAV vectors was comparable. It is clear that only the use of supernatant-derived vectors will considerably facilitate purification because of the absence or the presence of rather low concentrations of cell-derived contaminants.
Use of stable packaging & producer cell lines
Stable producer cell lines are cell lines containing the AAV functions rep and cap as well as the rAAV transgene construct with the transgene expression cassette flanked by the two ITRs. These cells are often based on HeLa cells [45], although A549 cells have also been used [47]. The development of these producer cell lines includes an intermediate step of a packaging cell line, which contains only the rep and cap functions of AAV, but not the rAAV transgene construct. Such cell lines have been developed either using HeLa or A549 cells leading, for instance, to the B-50 [48] and C12 [49] or K209 [50] cell lines, respectively. Finally, in order to establish the producer cell line, the packaging cells have to be transfected or transduced with a rAAV transgene plasmid or with a recombinant AAV vector, respectively. However, it should also be noted that some authors have developed the producer cell lines without the intermediate development of a packaging cell line. This can be achieved by the transfection of cells with a plasmid containing the rep-cap functions as well as a recombinant AAV transgene sequence [45,51,60].
Packaging cell lines

Figure 2. Packaging and producer cells of rAAV vectors. HeLa or A549 cells are transfected with a plasmid containing the rep2 (from AAV2)-capX (from any AAV serotype) sequences (Rep-Cap), which leads to establishment of packaging cells. When these cells are infected with adenovirus and 24h later by an E1-deleted adenovirus-AAV-Hyb(rid) virus (providing the rAAV transgene sequence), rAAV as well as adenovirus is produced. If the rAAV transgene sequence is stably integrated via plasmid transfection (pAAV-transgene) or vector transduction (rAAV-vector), the packaging cells become stable producer cells, which upon infection with adenovirus start to produce rAAV vector and adenovirus.
Since these cells contain the AAV functions rep and cap, they are also able to produce AAV vectors; however, in this case, they have to be sequentially infected with an adenoviral helper virus with functional genes of the E1 region and subsequently with an adenovirus-–AV hybrid virus (Ad-AAV Hyb. virus) containing the rAAV transgene sequence in the E1 region of the adenovirus [52]. Concerning the adenoviral helper virus, it is preferable that it is replication deficient to avoid its production and contamination of the final AAV vector product, although purification protocols have been developed and validated for the removal and inactivation of the adenoviral helper virus [45]. This replication deficiency was achieved by using an adenovirus with a functional E1 gene required to activate p5, p19 and p40 transcription units of the rep-cap genes and, in addition, containing a temperature sensitive mutation in E2B, for instance, for precluding contamination of the final rAAV preparation by the adenoviral helper virus [48]. Based on the understanding of underlying biological processes and factors influencing the generation of rAAV vectors, the following optimal production protocol has been developed [48,50,52]: the packaging cells are infected with a conditionally replication defective helper virus at a non-permissive temperature with an multiplicity of infection (MOI) dependent on which packaging cell line (HeLa- or A549-cell-based packaging line) is used. After 24 hours, the cells are superinfected with the adenovirus–AAV hybrid virus containing the gene of interest flanked by both ITRs at an MOI of 10 and the harvest is performed 48 hours later followed by purification. However, since the temperature-sensitive adenovirus mutants are prone to reversion and thus difficult to produce and to characterize, they have not been implemented for large-scale AAV production.
This production system is characterized by several advantages: in addition to five- to ten-fold improved levels in cell-specific vector production [54], it provides the real possibility for scale-up to industrial reactor scales when compared to the traditional transfection method. Another feature is its ability to generate replication-competent AAV (rcAAV)-free rAAV preparations (6 log10, respectively, adenovirus clearance [46]. Obviously, the use of replication-deficient adenovirus can alleviate this inconvenience [47]. However, these adenoviruses are sometimes characterized by instability [57] and lower rAAV vector titers in comparison to wild-type adenovirus. This was, for instance, observed by Jenny et al. when using a protease-deficient adenovirus (AdDPS) [59]. In the case of wild-type adenovirus, the optimal MOI ranges between 10 and 100 for ensuring amplification of rep and cap genes [55] as well as of the recombinant rAAV vector genome [53]. In this context, it should be noted that rAAV production levels are highly impacted by robust amplification of the AAV rep/cap genes upon adenovirus infection and not by the integrated vector plasmid copy number per se [60].
Optimal producer cell lines show cell-specific production levels in the range of 5 x 104 to 2 x 105 vg/cell [45,60] under serum-free suspension conditions. Furthermore, scale-up has been performed up to a 2000 L scale using stirred tank reactors [61] with the potential for generating up to 8 x 1016 vg (purified) per run. This production system is characterized by an absence of detectable rcAAV (below 0.0002% = below limit of detection), by a ratio of full-to-empty vector particles of ≥70% and a potency equivalent to rAAV vector produced via transient transfection [65].
For further information, in particular, on the underlying biology of AAV vector production in context of packaging and stable producer cell line, the discussion section of the article by Martin et al. [60] is recommended; in addition, the paper by Thorne et al. [61] has more information on issues of large-scale GMP production of rAAV vector and related regulatory issues when using stable producer cell lines.
Use of the recombinant HSV Type 1 expression system
HSV is another helper virus that can rescue and induce replication of AAV; thus this virus has also been envisaged for the production of rAAV vectors. This AAV production system is a pure infection-based system in which the non-modified production cells are infected by two different recombinant HSVs providing the AAV functions rep and cap as well as the rAAV vector with the transgene flanked by the two ITRs. In order to reduce or completely avoid the production of recombinant HSV (rHSV), which is highly pathogenic, this expression system is based on the use of ICP27-deficient rHSV helper virus, which is replication incompetent in normal cells. The ICP27-deficient rHSV virus can be propagated in V27 cells (recombinant Vero cells containing the essential UL54 gene encoding for and thus supplying ICP27 in trans) [62]. Furthermore, the two required AAV sequences (rep2/capX and the rAAV vector construct with the transgene cassette flanked by the two ITRs) are inserted into the (TK) locus of the ICP27-deficient HSV vector, each sequence cloned into a separate HSV vector [63](Figure 3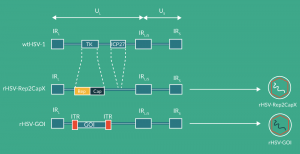
Initially, adherently grown HEK293 cells have been used for AAV production; however, to obtain the highest cell-specific AAV vector production (6500 ip/c), an MOI ratio of 12:2 (rHSV-rep2/cap2:rHSV-GFP) was required [65]. Staying with a CS10 system, infection conditions have been improved for the generation of AAV9 vectors. Although not a scalable process, this improvement allowed the generation of up to 2 x 1014 rAAV9 per CS10 due to a specific productivity of approximately 2 x 105 vg/c [66], which represents an increase of two- and five-fold, with respect to the non-optimized conditions and the HEK293-based transfection process, respectively.
Despite these improvements, the production conditions represent a drawback for large-scale production, because real scalability is ensured by suspension culture processes and low MOI is required to reduce the HSV vector amount per production run. This was improved by switching to BHK cells grown in suspension for which the MOI ratio could be reduced to 4:2 (rHSV-rep2/cap2 to rHSV-GFP) [67]. Optimal cell density for infection was established to be in the range between 1.6 x 106 and 3.2 x 106 c/ml and the optimal harvest time is 24 hours post-infection [67]. This process generates between 69000 ± 7500 and 11,3000 ± 16,400 DNase Resistant Particles (DRP = equivalent to vector genome [vg])/c at a 10 L scale (using WAVE reactors), which is equivalent to production yields obtained at spinner scale; average productivities of 2.4 x 1014 vg/L have been reported [67]. Scale-up has been performed to a 100 L reactor scale [68]. Moreover, it was shown that practically all assessed AAV serotypes could be produced with the HSV expression system: AAV1, AAV2 [65,67], AAV5, AAV8 [67], AAV9 [65].
Although replication-deficient HSV vectors are used for the production of AAV vectors, the generation of replication-competent HSV due to recombination events during production cannot be excluded wherefore the downstream processing protocol consisting of orthogonal purification principles must be validated for inactivation as well as removal of HSV. The established purification regime consisting of a detergent step, CIM Q followed by AVB affinity chromatography was able to clear at least 14.04 log10 of HSV [69]. Further advantages of this expression system is the absence of rcAAV generation and the in vivo vector potency of AAV1 in mice (DRP/IP ratio) being approximately five times higher when compared to AAV1 vectors generated by transient transfection [69].
More details on the development of the HSV expression system for the production of AAV vectors can be found in Clément et al [65].
Use of the baculovirus/insect cell system
A second infection-based production system for AAV vectors is the non-mammalian insect cell/baculovirus system, which is based on the use of mainly Sf9 cells (or their derivatives) grown in serum-free/animal-free medium in suspension culture allowing thus a very simple scale-up. This expression system was first developed by Urabe et al. [70]. They proposed the use of three different baculoviruses in which the three required functions were inserted into the classical polyhedrin locus. One baculovirus provided the cap-sequence under control of the very late baculoviral polyhedrin promoter in which the VP1 start codon ATG had been replaced by the less efficient ACG to reduce translational efficiency and thus allow the ribosomal machinery to scan down to the next low efficiency start codon ACG for VP2 expression and further down to the start codon ATG of VP3 for expression at high efficiency. The second baculovirus provided the AAV2 rep78 under control of a truncated version of the early promoter E1 of the baculovirus and rep52 under control of the late baculoviral p10 promoter, both in a head-to-head orientation. And finally, the third baculovirus provided the recombinant rAAV construct with the transgene cassette flanked by the ITRs (Figure 4A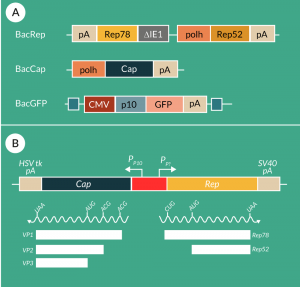
Smith et al. created the two baculovirus systems with the advantage of the reduction of the number of baculoviruses (to two) and tackling the instability issue of the rep-baculovirus by generating a unique sequence coding for rep78 and rep52 (Figure 4B). They mutated the start codon of the rep78 sequence into ACG and performed codon optimization for mutating the following nine in-frame ATGs into non-start codons; however, the ATG start codon of rep52 was retained [73]. With respect to the cap sequence, the only modifications consisted in codon optimization as carried out for the rep sequence. The rep and cap sequences driven by polyhedrin and p10 promoter, respectively, were put into a single baculovirus in a head-to-head position, with the rep cassette positioned in the clockwise lecture sense. Production levels of 7 x 104 vg/c have been reported; however, the expression levels of VP1 were still lower than for rAAV produced with the transient transfection system approach. Productions have been performed at a 200 L scale using the dual baculovirus system [74].
Chen kept three different baculoviruses; however, the rep and cap baculoviruses have been improved. He inserted an artificial intron harboring the polyhedrin promoter into the rep78 coding sequence at the p19 promoter region in order to express both rep proteins (78 and 52) from a single rep coding sequence. A similar solution was used for the cap baculovirus, meaning that an intron as used for the rep construct was inserted into the VP1 sequence upstream of the VP2 start codon. The original start codon (ATG) of VP1 was kept and ensured high expression levels of VP1, which transduced to two-fold higher AAV vector potency than when using ACG as start codon for VP1 (Figure 4C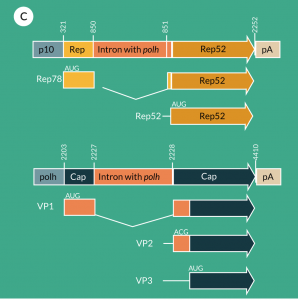
Hermens et al. used a single rep-construct by replacing the original ATG start codon of rep78 by the suboptimal initiation ACG effecting partial exon skipping and thus expression of downstream rep52 protein [77]. Expression was controlled by a polyhedrin promoter. This construct stayed stable for at least five passages. As also carried out by Chen, the suboptimal expression of the VP1 protein was increased by the modification of the start codon (CTG instead of ACG) leading to VP1 expression levels as found for the traditional plasmid transfection system [78]. As for Chen [75], the triple baculovirus system was kept.
Production issues & further improvements
Although the insect cell baculovirus system is scalable, can produce in principle unlimited amounts of rAAV and is routinely used for the GMP production of vector lots for clinical as well as for commercial use, further improvements are required to ameliorate vector titer, quality and potency:
Increase in AAV vector titer
In the originally developed production system, Sf9 cells are infected at a cell density of 1–2 x 106 c/ml [70]. In principle, it can be stated that at constant specific production rate the final vector titer is directly related to the cell concentration in a given cell culture system. This would signify that a simple increase in the cell concentration should lead to a corresponding increase in vector titer. However, as for many other viral systems, the baculovirus expression system is also characterized by the so-called ‘cell density effect’ [79–83], which implies that beyond a certain cell density, a further increase in cell concentration does not lead to further increase in vector titer. This can be explained by lack of certain essential medium components, because Mena et al. could show that the feeding of high-density cultures 24 hours before, at and 24 hours after virus infection with a medium concentrate could maintain the specific vector production rate up to an infection cell density of 10 x 106c/ml [84]. However, it should be noted that at these cell numbers the infections can only be performed with low MOIs (e.g., 0.05 per baculovirus) because at high MOIs, the culture would be too much diluted with spent medium from the baculovirus production culture. However, it is important to note that only 60–70% of the cells will be infected with the two different viruses [85], meaning that part of the cells will not produce functional rAAV vectors.
AAV vector quality & potency
Different biological parts of the insect cell/baculovirus system could probably be optimized for enhancing vector quality and potency. With respect to the choice of the ITRs, Dickx et al. [86] could show that the use of complete wild-type ITRs (FLIP – FLOP) instead of the use of truncated versions of FLOP ITRs (SUB201-ITRs) largely used without negative impact in context of the transient transfection-based production system [87-89] led to an increase in the full-to-empty particle ratio (increase from 10 to 40%) and a reduced (approximately ten-fold) encapsidation of baculovirus DNA sequences. Noordman et al. showed that the encapsidation of baculoviral DNA sequences could be reduced via mutation of the rep-DNA sequence when producing rAAV5 vectors [90]. These mutations led to three- to five-fold, five- to 13-fold, and 25–52-fold reduction in encapsidation of left ORF (603), right ORF (1629) and HR3 residual baculoviral DNA, respectively. In parallel, the use of some of these mutated rep proteins led to increased vector titers.
Baculoviruses contain the cathepsin gene (a cysteine protease), which works in tandem during baculovirus infection of lepidopteran larvae to achieve dissemination of the occluded form of the baculovirus progeny [91,92]. Though this is important for the wild-type virus, the presence of an active protease during the production of recombinant proteins can be a drawback. In this context, Galibert et al. demonstrated that the VP1 of certain AAV serotypes (1, 6, 8) is cleaved during vector production whereas this was not observed for other serotypes (2, 9, rh10) [93]. Either use of the cathepsin-specific protease inhibitor E64 [91] or the disruption of the cathepsin gene led to AAV8 vector preparations with intact VP1 capsid proteins. It was shown that the in vitro and in vivo infectivity of the AAV8 vectors was two- to four-fold increased in comparison to those produced using baculoviruses with an intact cathepsin gene. Although these are only some of the recent developments for improving the Sf9/baculovirus system for AAV production, there are a lot of further possible improvements to be made; this concerns in particular, the timely regulation of the expression of the AAV rep and cap genes and the baculovirus helper functions.
An additional reduction in the number of baculoviruses leads to a single baculovirus system
The initial baculovirus system as published by Urabe et al. (2002) was based on three different baculoviruses [70], which could be reduced to two baculoviruses [73]. However, the ultimate improvement and in the same time simplification from a user’s point of view consists in the use of a single baculovirus for inducing AAV vector production (at low MOI all cells would receive the whole set of genes for producing AAV vectors – compare with Rivollet et al.[85]). Two approaches have been developed for achieving this objective:
MONOBAC system
The first approach is the MONOBAC system, which is based on the fact that several of the genes of a baculovirus bacmid are transcriptionally very active but not essential for in vitro use of the baculovirus. Thus we have decided to place the rep-cap construct developed by Smith et al. [73] into the EGT locus and kept the classical polyhedrin locus for the insertion of the recombinant AAV vector cassette (the transgene cassette flanked by the two ITRs) (Figure 4D
OneBac system
The second single baculovirus system is the OneBac system whose first generation was published by Aslanidi et al [96]. The principle of this system is relatively simple. The producer cell line (Sf9) harbors stably integrated the rep and cap functions, which are amplified and expressed upon infection with the recombinant baculovirus providing the rAAV genome. The rep and cap expression cassettes have been constructed using an identical approach: they consist of homologous region (hr) 2 followed by the AAV rep binding element (RBE) a stuffer sequence and finally the rep or cap sequence, the expression of both driven by the very late baculovirus polyhedrin promoter. The expression is only induced after infection with baculovirus via the binding of the immediate-early trans-regulator 1 (IE-1) to the hr2 target sequence inducing expression via a combination of the amplification of integrated resident genes (up to 1200 copies per cell) and the enhancement of the expression. Furthermore, the expression of rep78 leads to the boosting of the integrated genes (via integration with the RBE). This system leads to a ten-fold increase in AAV vector production in comparison to the original baculovirus system – up to a specific production of 5 x 105 vg/c (rAAV2) exceeding typical yields of current rAAV production systems. However, it could also be established that probably due to this amplification a considerable percentage of AAV particles containing rep and cap sequences were detected (our own unpublished results, [97]). Thus, this first generation could not be further improved for an eventual GMP application. Mietzsch et al [97] developed the second-generation OneBac system (OneBac 2.0), in which in the first line the RBE element responsible for the encapsidation of high frequencies of rep and cap sequences into AAV capsids was removed (Figure 4E
General ‘pseudotyping’ issues
In the vast majority of studies a pseudotyping strategy has been adopted for simplicity reasons, meaning that only the cap sequence was from a desired serotype, whereas the ITRs as well as the rep sequences were derived from AAV2 [100]. Although this represents a considerable simplification of production of different AAV serotypes because of the simple switch from one capsid sequence to another one, it is not necessarily the best way to produce high vector amounts of elevated quality. In the context of the optimization of the quality and quantity of AAV vector production, Ling et al. generated AAV3b vectors using a completely homologous expression system based on the use of rep protein and ITRs sequences derived from AAV3b [101]. They could show that when using this homologous expression system (in a transient transfection context) four-fold higher titers as well as four-fold higher transduction efficiencies of a human hepatocellular cell line in vitro were obtained in comparison to the traditional heterologous system characterized by the use of rep and ITR sequences derived from AAV2. Thus it could be inferred that the use of rep and ITR sequences homologous to the capsid serotype might also be a way to improve vector titers and efficacy of other AAV serotypes.
Comparison of different production systems
Relatively few comparisons have been performed between AAV vectors produced with the different production systems. Based on published data, Table 1 compares the different production systems (excluding the packaging cell line-based system because up to now no real scale-up has been performed for this system) with respect to specific production rate, the largest production scale established up to now, the vector amount produced per culture litre using standard conditions as well as some safety parameters, including the generation of rcAAVs, the percentage of full vector particles before purification and the encapsidation of rep and cap sequences as far as communicated.
| Table 1. Comparison of different rAAV production systems with respect to specific vector production rate, volumetric production level, largest scale established as well as several safety aspects (generation of rcAAV, ratio of full to empty particles, level of encapsidated rep, cap, and HSV DNA). | ||||||||
|---|---|---|---|---|---|---|---|---|
| Production system | Biological system used | Cell specific production rate (vg/c) | Volumetric production (vg/L) | Largest scale used | rcAAV production | Percentage of full particles (%) | Encapsidated rep/cap, HSV sequences (%) | Ref. |
| Plasmid transfection | Selected suspension adapted HEK293 clone | 1-2x105 (AAV2) | 1014 | 20L (WAVE)** | +* | 5-50 | Rep: 0.3-1.5 [33] Cap:0.4-1 [130] ; 0.016-0.024 [98] | [28] |
| Stable cell line | HeLa (rep-cap/rAAV vector), infection with wt adenovirus 5 | >5x104*** | >5x1013 | 250L (STR) -> 2000L (STR) | Below detection level | >50 | [45], [61], [60] | |
| >105 | Shake flasks** | <0.0002% | >70 | 0.02-0.05 (rep, cap) | ||||
| Herpes simplex type 1 | Suspension adapted BHK cells, infection with 2 viruses (rHSV-rep2capX/rHSV-rAAV vector), MOI=4/2 | 6.9x104 1.13x105 | 2.4x1014 (AAV1) | 25L (WAVE) -> 100L (WAVE) | Below detection level | 97.55±0.24% [131] | HSV: 0.007-0.012 | [63], [67], [64] |
| Baculovirus system | Sf9, infection with 2 viruses (Bac-rep2/capX/Bac-rAAV-vector), MOI=0.05/0.05 1.6/1.6 | 104 105 | 9.4x1013 (AAV1) | 200L (STR, WAVE) | Below detection level | 10-40%**** (AAV8)[86] | Cap: 0.016 | [12], [74] |
| 8.3x1013 (AAV6) | >70% (AAV2) [103] | Rep: 0.019-0.024 [76] | ||||||
| OneBac 2.0 system | Stable Sf9 cell line (rep-cap), infection with 1 virus (BAC-rAAV vector), MOI=5 | ~105 (AAV5) | 1.4x11515 | Small scale** | Below detection level | No information | Cap: 0.02 Rep: <0.001 | [97] |
| *Depending on the plasmid system used (compare with Emmerling et al. [23]). **Large production scale possible. ***Selection criteria. ****Depending on the ITR construct | ||||||||
With respect to the specific vector yield per cell, all expression systems as well as the transfection system-based on a selected HEK293 cell clone adapted to suspension growth are comparable and the specific yield ranges between 5 x 104 and 2 x 105 vg/c. For all production systems it can be stated that real scale-up is only possible when using a suspension culture. However, this has to be relativized for the HEK293(T) cell-based transfection process for which also an adherent production process using a fixed bed reactor (e.g., iCELLis® reactor) might be used. Furthermore, it has to be indicated here that fixed bed reactors are also well adapted to viral infection based production systems [20,102] although not yet shown for AAV production.
In principle, all production systems generate vector titers ranging from 5 x 1013 to 2.4 x 1014 vg/L or DRP/L. An exception in this context seems to be the baculovirus system, for which different production levels have been reported for various AAV serotypes. While for the production of AAV2 and AAV7m8, similar production levels have been reported for the baculovirus system and the HEK293 cells-based transfection system; production levels for AAVrh10 were approximately two times lower for the baculovirus system [103]. In the specific case of AAV8, the baculovirus system underperformed by a factor of 5–10x [104] or generated non-functional AAV8 vector capsids devoid of VP1 when produced at large scale (200 L) [105]. These observations clearly infer that in the case of the baculovirus expression system no generalized view is permitted, and production and functionality issues have to be assessed for a chosen serotype.
Attention has to be paid to the fact that the differences in the production levels (when comparing different production systems) can be explained by different production cell densities and specific production rates. However, it should also be kept in mind that the titration methods were not harmonized/standardized making a real comparison challenging.
With respect to genetic stability, the used biological raw materials have to show stability exceeding that required for the whole process. This concerns the producer cells as well as viral seed stocks. In this context, genetic stability of HeLa-based stable producer cell lines has been demonstrated for more than 60 population doublings [45] and the recombinant viruses for the HSV and the baculovirus-based production systems have been shown to be stable for 13 [106] and seven [73] successive passages, respectively. Furthermore, no generation of rcAAV has ever been reported for all expression systems (below limit of detection of the assay system used) except for the transfection based production process for which recombination events can generate rcAAV [107].
From a practical point of view, all suspension-based productions can be performed either in WAVE-type or stirred tank reactors, with a certain advantage for the stirred tank reactor due to the easier scale-up (which is theoretically unlimited) as well as the possibility to infect cultures at higher cell densities than 106 c/mL due to the better mass transfer features of stirred tank reactors. While the maximal scale of the WAVE reactor is limited to 200–250 L, because at larger volumes, mass transfer becomes critical and thus limiting, stirred tank reactors for animal cells have been scaled up to reactor sizes with 20,000L working volume [108]. Today reactor scales of 200–250 L are routinely used for the production of clinical-grade AAV vectors and the use of a 2000 L reactor scale is under implementation [45,61]. It should be added here, that the HSV and the baculovirus-based production systems should in principle also be scalable although the modification of the physico-chemical parameters (e.g., shear stress) and their impact on cellular metabolism during scale-up to larger scales merits consideration wherefore WAVE type reactor systems might be more adapted.
Side-by-side comparisons of different AAV expression systems with respect to in vivo transduction efficiency are rarely performed. In this context, Zhang et al. compared the efficiency of transduction of striatum and cortex tissue by AAV2 vectors produced with the herpes simplex, the baculovirus system (as developed by Chen [75]), and the HEK293-based transfection process [109]. Similar transduction efficiencies as well as cellular tropism were observed for the AAV2 vector particles produced with the three expression systems, signifying that the different production systems are apparently able to generate AAV vectors of comparable efficiency. By comparing different AAV serotypes (2, 7m8, rh10) Ramirez et al. could also show that these serotypes produced with the baculovirus expression system (as developed by Chen [75]) or via triple transfection of HEK293 cells had comparable in vitro and in vivo potency [103]. With respect to AAV vector particles generated using the HSV expression system, Kang et al. and Adamson-Small et al. showed that the potency of HSV expression system derived AAV particles was superior to that of AAV particles produced via a transfection-based process [65,66]. This concerned AAV serotypes 1 and 9, respectively.
These comparisons infer very clearly that at this moment it is impossible to provide a clear statement on the quality and capacity of a given AAV expression system versus another system because insufficient data available for comparisons and in most cases a direct comparison is not possible because different serotypes and vector constructs have been used not allowing a real comparison.
Purification
There are several means for purifying AAV vectors, starting with traditional density gradient ultracentrifugation using CsCl or iodixanol [110,111] going up to the novel affinity chromatography gels initially developed by BAC/GE Healthcare (AVB chromatography) and further improved by Invitrogen for specific AAV serotypes (POROS CaptureSelect AAV8 Affinity Matrix and POROS CaptureSelect AAV9 Affinity Matrix). Different aspects of AAV purification have been described previously (e.g., [111,112]) and do not need another repetition. Therefore only two issues concerning downstream processing of AAV vectors are discussed here.
Availability & implementation of novel affinity chromatography matrices
The appearance of novel affinity matrices specific for AAV8 and AAV9 have allowed the development of a highly efficient and vector specific purification protocols consisting of clarification, affinity chromatography, tangential flow filtration and eventually a gel filtration step for formulation purposes. AVB column chromatography was initially developed for the affinity purification of AAV2 [125] and was reported by the manufacturer to capture AAV serotypes 1, 2, 3 and 5. Recently, Mietzsch et al [99] communicated that AAV1-8, AAVrh.10 and AAV12 could be purified from crude cell lysate, whereas this was not possible for AAV9 and AAV11. Later, Wang et al. established that AVB affinity chromatography column had a high affinity towards AAV3B, AAVrh.10 and AAVhu.37 and a low affinity/absence of affinity towards AAV8, rh.64R1 and AAV9 [128]. Since in particular for the purification of AAV8 and AAV9 vectors, AVB chromatography was not or only partially adapted and since both serotypes are of high interest for clinical use, two new matrices with specific llama antibody ligands (POROS Capture Select Affinity Matrix) have been developed. In the case of purification of AAV8 vectors, this novel support required an approximately ten-fold reduced support volume due to the increased ligand density and led to an increase in the overall yield from 20–30% to approximately 60%. A similar overall recovery yield was communicated for AAV9 vectors [126]. This represents an interesting improvement with respect to cost of goods and operator time per run.
One issue related to the use of affinity chromatography based on the use of these new affinity ligands is the fact that full particles cannot be separated from empty particles, meaning that additional steps, including ion-exchange chromatography or ultracentrifugation, are required when empty AAV particles have to be removed (see below).
Full–empty AAV particle issue
A specific issue is the presence of empty particles in vector preparations, which are almost always co-produced. In the case of the transient transfection-based production system, empty capsids are produced at a high frequency going up to 50–95% of the total particle count [113]. In order to separate empty from full AAV particles, the Center for Cellular and Molecular Therapeutics at the Children Hospital of Philadelphia used a downstream processing protocol including an ion-exchange chromatography as capture step followed by a CsCl ultracentrifugation for manufacturing of GMP vector lots for clinical studies [114]. For instance, this protocol was used for generation of vector preparations used for the different clinical trials performed in Philadelphia: ssAAV2-FactorIX for hemophilia B treatment [115] or AAV2-hRPE65v2 for treatment of Leber’s congenital amaurosis [116,117]. On the other hand, the vector preparation used for the scAAV8-FactorIX trial contained an elevated level of empty particles, which was estimated to exceed the full particles ten-fold [1]. In both cases, efficient vector transduction was observed although only transiently for the hemophilia B trial performed with ssAAV2 vectors free of empty capsids. Thus to date, vector preparations containing only full vector particles on one side and preparations with mostly empty particles (10% of full particles) have been used for clinical studies. With respect to the presence of a percentage of empty AAV vector capsids it could be shown that the presence of empty vector particles was beneficial for neutralizing anti-AAV antibodies present, which would completely block transduction. This absorbance happened in a dose-dependent manner [118]. However, on the other hand, Gao et al. showed in two mouse models, that the presence of empty vector particles had a negative impact on liver transduction in a dose-dependent manner [119]. The conclusions from both studies are the following: the vector preparation should be devoid of empty particles for avoiding the competitive situation of empty and full particles for the same target. However, as shown by Mingozzi et al. [118] the presence of empty capsids can be beneficial for absorbing eventually present anti-AAV antibodies. Thus in order to avoid this competitive situation, they could show that use of a mutant AAV capsid able to interact with the anti-AAV antibodies but unable to interact with the cellular receptor could be a solution: removal/absorption of anti-AAV antibodies without competing for the cellular target sites of the functional AAV vector with the transgene to be transferred. In principle, such an approach can be developed via the use of AAV preparations, which contain only full particles to which a certain percentage of these AAV particles with a mutant capsid have been added.
As mentioned, at research scale, the separation of full and empty AAV particles is easily performed via CsCl [110,111] or iodixanol [120,121] gradient centrifugation; whereby iodixanol is preferred because it is non-toxic – thus no extensive dialysis is required as when CsCl is used, and iodixanol prevents aggregation of rAAV particles and does not reduce infectivity [120,127].
Although this separation step can be used at later stages of purification schemes, it is not a scalable purification step. Apart from the use of production methods reducing or avoiding the production of empty particles, the only other way to separate full from empty particles is the use of column chromatography. Since it could be established that the isoelectric point of empty particles is significantly higher than of full particles, probably due to the absence of DNA in the empty particles [124], it is possible to separate them using ion-exchange chromatography. This was shown by Davidoff et al. [123] and by Okada et al. [124] who developed a two stage ion-exchange chromatography process based on a strong cation-exchange column (Mustang S), which retains only empty particles whereas full particles are found in the flow through, this is followed by a strong anion-exchange column (Mustang Q), which retains full AAV particles, which can then be eluted using a salt gradient/step gradient. Using this method, Davidoff et al. [123] generated AAV8 vector preparations with only 5–10% of residual empty vector capsids, whereas Okada et al. [124] used a similar protocol for the purification of AAV1 vectors contaminated only by 0.8% of empty particles. Finally, based on the same principle – differences in the isoelectric point of fullet al. also used a two-step protocol with a first cation-exchange chromatography, however, which retains full as empty particles [122]. The eluted material was charged to a Q-Sepharose anion exchanger to separate full from empty particles at elevated pH using a very shallow salt gradient. Using this protocol, the empty particle load could be reduced by 86-fold with approximately 20% of residual empty particles in the final vector preparation. This method was also able to separate empty particles of AAV6. The separation of empty form full particles of different serotypes is feasible but has to be optimized by selecting different elution conditions, choice of salt and type of resins. It should also be mentioned here that to date, nobody has shown a large-scale application of this separation method.
Translational insight
Different scalable manufacturing systems have been developed for the large-scale production of AAV vectors for research, development, clinical evaluation and routine use for the treatment of rare diseases. Although most of the production-related issues have been settled today (e.g., large-scale production is possible using different biological systems), there are still several issues that need solutions. These are the large-scale production of vector preparations free of empty AAV particles including scalable separation methods for removing empty AAV particles, the question of the treatment of patients who are positive for anti-AAV antibodies because of a previous AAV infection or previous treatment with AAV vectors, as well as the increase of vector potency, which is generally low. An improvement in vector potency will have a direct impact on the vector dose to be administered and thus also on the vector quantities needed as, for instance, could be shown for AAV vectors with an improved percentage of functional VP1 protein (e.g., [93]). A further improvement of the AAV production will consist in the establishment of the routine use of homologous AAV systems since it has been shown that the use of such an homologous system leads to titer and potency increase [101]. Furthermore, the implementation of next-generation sequencing will provide a great deal of information on the packaged vector genome, as well as on the encapsidated DNA sequences from different origins (cellular origin, helper virus origin) (e.g., [129]) and will be a crucial tool for process development as well as optimization purposes. At the end of the day, these and further improvements will lead to more efficient production processes and a high-quality vector system with improved biosafety.
Financial & competing interests disclosure
The author has no relevant financial involvement with an organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock options or ownership, expert testimony, grants or patents received or pending, or royalties. No writing assistance was utilized in the production of this manuscript.
References
1. Nathwani AC, Tuddenham EGD, Rangarajan S et al. Adenovirus-associated virus vector–mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011; 365(25): 2357–65.
https://doi.org/10.1056/NEJMoa1108046
2. Nathwani AC, Reiss UM, Tuddenham EGD et al. Long-Term Safety and Efficacy of Factor IX Gene Therapy in Hemophilia B. N. Engl. J. Med. 2014; 371(21): 1994–2004.
https://doi.org/10.1056/NEJMoa1407309
3. Nathwani AC, Nienhuis AW, Davidoff AM. Our journey to successful gene therapy for hemophilia B. Hum. Gene Ther. 2014; 25(11): 923–6.
https://doi.org/10.1089/hum.2014.2540
4. Patel N, Reiss U, Davidoff AM et al. Progress towards gene therapy for haemophilia B. Int. J. Hematol. 2014; 99(4): 372–6.
https://doi.org/10.1007/s12185-014-1523-0
5. Cideciyan AV, Jacobson SG, Beltran WA et al. Human retinal gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc. Natl. Acad. Sci. USA 2008; 105(39): 15112–7.
https://doi.org/10.1073/pnas.0807027105
6. Maguire AM, Simonelli F, Pierce EA et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N. Engl. J. Med. 2008; 358(21): 2240–8.
https://doi.org/10.1056/NEJMoa0802315
7. Bainbridge JW, Smith AJ, Barker SSet al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N. Engl. J. Med. 2008; 358(21): 2231–9.
https://doi.org/10.1056/NEJMoa0802268
8. Hauswirth WW, Aleman TS, Kaushal S et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: shortx-term results of a phase I trial. Hum Gene Ther. 2008; 19(10): 979–90.
https://doi.org/10.1089/hum.2008.107
9. Spark Therapeutics. Spark Therapeutics announces positive top-line results from pivotal Phase 3 Trial of SPK-RPE65 for genetic blinding conditions. Globe Newswire, Philadelphia, PA, October 2015.
10. Ylä-Herttuala S. Endgame: Glybera finally recommended for approval as the first gene therapy drug in the European union. Mol. Ther. 2012; 20(10): 1831–2.
https://doi.org/10.1038/mt.2012.194
11. Allay JA, Sleep S, Long S et al.. Good manufacturing practice production of self-complementary serotype 8 adeno-associated viral vector for a hemophilia B clinical trial. Hum. Gene Ther. 2011; 22(5): 595–604.
https://doi.org/10.1089/hum.2010.202
12. Merten O-W, Moullier P. Approaches to large scale production of AAV vectors. In: Clinibook: Clinical gene transfer – state-of-the-art. Cohen-Haguenauer O (Ed.). Editions EDK/Groupe EDP Sciences, Paris, 71–82 (2012).
13. Le Guiner C, Montus M, Servais L et al. Forelimb treatment in a large cohort of dystrophic dogs supports delivery of a recombinant AAV for exon skipping in Duchenne patients. Mol. Ther. 2014; 22(11): 1923–35.
https://doi.org/10.1038/mt.2014.151
14. Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 1998; 72(3): 2224–32
15. Matsushita T, Elliger S, Elliger C et al. Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther. 1998; 5(7): 938–45.
https://doi.org/10.1038/sj.gt.3300680
16. Galibert L, Merten O-W. Latest developments in the large-scale production of adeno-associated virus vectors in insect cells toward the treatment of neuromuscular diseases. J. Invertebr. Pathol. 2011; 107: S80–S93.
https://doi.org/10.1016/j.jip.2011.05.008
17. Grimm D, Kay MA, Kleinschmidt JA. Helper virus-free, optically controllable, and two-plasmid-based production of adeno-associated virus vectors of serotypes 1 to 6. Mol. Ther. 2003; 7(6): 839–50.
https://doi.org/10.1016/S1525-0016(03)00095-9
18. Wright JF, Wellman J, High KA. Manufacturing and regulatory strategies for clinical AAV2-hRPE65. Curr. Gene Ther. 2010; 10(5): 341–9.
https://doi.org/10.2174/156652310793180715
19. Lock M, Alvira M, Vandenberghe LH et al. Rapid, simple, and versatile manufacturing of recombinant adeno-associated viral vectors at scale. Hum. Gene Ther. 2010; 21(10): 1259–71.
https://doi.org/10.1089/hum.2010.055
20. Merten OW, Cruz PE, Rochette C et al. Comparison of different bioreactor systems for the production of high titer retroviral vectors. Biotechnol Prog. 2001; 17(2): 326–35.
https://doi.org/10.1021/bp000162z
21. Wang X, Olszewska M, Qu J et al.. Large-scale clinical-grade retroviral vector production in a fixed-bed bioreactor. J. Immunother. 2015; 38(3): 127–35.
https://doi.org/10.1097/CJI.0000000000000072
22. Powers AD, Piras BA, Clark RK et al. Development and Optimization of AAV hFIX Particles by Transient Transfection in an iCELLis® Fixed-Bed Bioreactor. Hum. Gene Ther. Methods 2016; 27(3): 112–21.
https://doi.org/10.1089/hgtb.2016.021
23. Emmerling VV, Pegel A, Milian EG et al. Rational plasmid design and bioprocess optimization to enhance recombinant adeno-associated virus (AAV) productivity in mammalian cells. Biotechnol. J. 2016; 11(2): 290–7.
https://doi.org/10.1002/biot.201500176
24. Reniers A, van Huffel C, Otto-Wilhelm Merten O-W et al.. Development of a scalable viral vector upstream process for gene therapy: rAAV-8 production by transient transfection of HEK-293 cells in iCELLis™ bioreactor. Poster at the ESGCT Meeting, Florence, Italy, 18–21 Oct 2016.
25. Pham PL, Kamen A, Durocher Y. Large-scale transfection of mammalian cells for the fast production of recombinant protein. Mol. Biotechnol. 2006; 34(2): 225–37.
https://doi.org/10.1385/MB:34:2:225
26. Durocher Y, Pham PL, St-Laurent G et al.. Scalable serum-free production of recombinant adeno-associated virus type 2 by transfection of 293 suspension cells. J. Virol. Methods 2007; 144(1–2): 32–40.
https://doi.org/10.1016/j.jviromet.2007.03.014
27. Chahal PS, Schulze E, Tran R et al. Production of adeno-associated virus (AAV) serotypes by transient transfection of HEK293 cell suspension cultures for gene delivery. J. Virol. Methods 2014; 196: 163–73.
https://doi.org/10.1016/j.jviromet.2013.10.038
28. Grieger JC, Soltys SM, Samulski RJ. Production of recombinant adeno-associated virus vectors using suspension HEK293 cells and continuous harvest of vector from the culture media for GMP FIX and FLT1 clinical vector. Mol. Ther. 2016; 24(2): 287–97.
https://doi.org/10.1038/mt.2015.187
29. Stacey GN, Merten O-W. Host cells and cell banking. Meth. Mol. Biol. 2011; 737: 45–88.
https://doi.org/10.1007/978-1-61779-095-9_3
30. Clément N, Grieger JC. Manufacturing of recombinant adeno-associated viral vectors for clinical trials. Mol. Ther. Meth. Clin. Dev. 2016; 3: 16002.
https://doi.org/10.1038/mtm.2016.2
31. Bauer S, Kirschning CJ, Häcker H et al. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc. Natl Acad. Sci. USA 2001; 98(16): 9237–42.
https://doi.org/10.1073/pnas.161293498
32. Hyde SC, Pringle IA, Abdullah S et al. CpG-free plasmids confer reduced inflammation and sustained pulmonary gene expression. Nat. Biotechnol. 2008; 26(5): 549–51.
https://doi.org/10.1038/nbt1399
33. Chadeuf G, Ciron C, Moullier P et al. Evidence for encapsidation of prokaryotic sequences during recombinant adeno-associated virus production and their in vivo persistence after vector delivery. Mol. Ther. 2005; 12(4): 744–53.
https://doi.org/10.1016/j.ymthe.2005.06.003
34. Schnödt M, Schmeer M, Kracher Bet al.. DNA Minicircle technology improves purity of adeno-associated viral vector preparations. Mol. Ther. Nucleic Acids 2016; 5: e355.
https://doi.org/10.1038/mtna.2016.60
35. Kobelt K, Schleef M, Schmeer M et al. Performance of high quality minicircle DNA for in vitro and in vivo gene transfer. Mol. Biotechnol. 2013; 53(1): 80–9.
https://doi.org/10.1007/s12033-012-9535-6
36. Mayrhofer P, Blaesen M, Schleef M et al. Minicircle-DNA production by site specific recombination and protein-DNA interaction chromatography. J. Gene Med. 2008; 10(11): 1253–69.
https://doi.org/10.1002/jgm.1243
37. Grimm D, Kern A, Rittner K et al. Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum. Gene Ther. 1998; 9(18): 2745–60.
https://doi.org/10.1089/hum.1998.9.18-2745
38. Grimm D, Kay MA, Kleinschmidt JA. Helper virus-free, optically controllable, and two-plasmid-based production of adeno-associated virus vectors of serotypes 1 to 6. Mol. Ther. 2003; 7(6): 839–50.
https://doi.org/10.1016/S1525-0016(03)00095-9
39. Giacca M, Zacchigna S. Virus-mediated gene delivery for human gene therapy. J. Control. Release 2012; 161(2): 377–88.
https://doi.org/10.1016/j.jconrel.2012.04.008
40. Jenny C, Toublanc E, Danos O et al. Evaluation of a Serum-free Medium for the Production of rAAV-2 using HeLa Derived Producer Cells. Cytotechnology 2005; 49(1): 11–23.
https://doi.org/10.1007/s10616-005-5361-z
41. Vandenberghe LH, Xiao R, Lock M et al. Efficient serotype-dependent release of functional vector into the culture medium during adeno-associated virus manufacturing. Hum. Gene Ther. 2010; 21(10): 1251–7.
https://doi.org/10.1089/hum.2010.107
42. Benskey M, Sandoval IM, Manfredsson FP. Continuous Collection of Adeno-Associated Virus from Producer Cell Medium Significantly Increases Total Viral Yield. Hum. Gene Ther Methods 2016; 27(1): 32–45.
https://doi.org/10.1089/hgtb.2015.117
43. Doria M, Ferrara A, Auricchio A. AAV2/8 vectors purified from culture medium with a simple and rapid protocol transduce murine liver, muscle, and retina efficiently. Hum. Gene Ther. Methods 2013; 24(6): 392–8.
https://doi.org/10.1089/hgtb.2013.155
44. Lock M, Alvira M, Vandenberghe LH et al. Rapid, simple, and versatile manufacturing of recombinant adeno-associated viral vectors at scale. Hum. Gene Ther. 2010; 21(10): 1259–71.
https://doi.org/10.1089/hum.2010.055
45. Thorne BA, Takeya RK, Peluso RW. Manufacturing recombinant adeno-associated viral vectors from producer cell clones. Hum. Gene Ther. 2009; 20(7): 707–14.
https://doi.org/10.1089/hum.2009.070
46. Thorne BA, Quigley P, Nichols, G et al. Characterizing clearance of helper adenovirus by a clinical rAAV1 manufacturing process. Biologicals 2008; 36(1): 7–18.
https://doi.org/10.1016/j.biologicals.2007.04.001
47. Farson D, Harding TC, Tao L et al. Development and characterization of a cell line for large-scale, serum-free production of recombinant adeno-associated viral vectors. J. Gene Med. 2004; 6(12): 1369–81.
https://doi.org/10.1002/jgm.622
48. Gao GP, Qu G, Faust LZ et al. High-titer adeno-associated viral vectors from a Rep/Cap cell line and hybrid shuttle virus. Hum. Gene Ther. 1998; 9(16): 2353–62.
https://doi.org/10.1089/hum.1998.9.16-2353
49. Clark KR, Voulgaropoulou F, Johnson PR. A stable cell line carrying adenovirus-inducible rep and cap genes allows for infectivity titration of adeno-associated virus vectors. Gene Ther. 1996; 3(12): 1124–32.
50. Gao GP, Lu F, Sanmiguel JC et al. Rep/Cap gene amplification and high-yield production of AAV in an A549 cell line expressing Rep/Cap. Mol. Ther. 2002; 5(5 Pt 1): 644–9.
https://doi.org/10.1006/mthe.2001.0591
51. Clark KR, Voulgaropoulou F, Fraley DM et al. Cell lines for the production of recombinant adeno-associated virus. Hum. Gene Ther. 1995; 6(10): 1329–41.
https://doi.org/10.1089/hum.1995.6.10-1329
52. Liu XL, Clark KR, Johnson PR. Production of recombinant adeno-associated virus vectors using a packaging cell line and a hybrid recombinant adenovirus. Gene Ther. 1999; 6(2): 293–9.
https://doi.org/10.1038/sj.gt.3300807
53. Liu X, Voulgaropoulou F, Chen R et al. Selective Rep-Cap gene amplification as a mechanism for high-titer recombinant AAV production from stable cell lines. Mol. Ther. 2000; 2(4): 394–403.
https://doi.org/10.1006/mthe.2000.0132
54. Zhang H, Xie J, Xie Q et al. Adenovirus-adeno-associated virus hybrid for large-scale recombinant adeno-associated virus production. Hum. Gene Ther. 2009; 20(9): 922–9.
https://doi.org/10.1089/hum.2009.125
55. Chadeuf G, Favre D, Tessier J et al. Efficient recombinant adeno-associated virus production by a stable rep-cap HeLa cell line correlates with adenovirus-induced amplification of the integrated rep-cap genome. J. Gene Med. 2000; 2(4): 260–8.
https://doi.org/10.1002/1521-2254(200007/08)2:43.0.CO;2-8
56. Matthews LC, Gray JT, Gallagher MR et al. Recombinant adeno-associated viral vector production using stable packaging and producer cell lines. Methods Enzymol. 2002; 346: 393–413.
https://doi.org/10.1016/S0076-6879(02)46068-5
57. Blouin V, Brument N, Toublanc E et al. Improving rAAV production and purification: towards the definition of a scaleable process. J. Gene Med. 2004; 6(Suppl. 1): S223–8.
https://doi.org/10.1002/jgm.505
58. Toublanc E, Benraiss A, Bonnin D et al. Identification of a replication-defective herpes simplex virus for recombinant adeno-associated virus type 2 (AAV2) particle assembly using stable producer cell lines. J. Gene Med. 2004; 6(5): 555–64.
https://doi.org/10.1002/jgm.542
59. Jenny C, Toublanc E, Danos O et al. Evaluation of a Serum-free Medium for the Production of rAAV-2 using HeLa Derived Producer Cells. Cytotechnology 2005; 49(1), 11-23.
https://doi.org/10.1007/s10616-005-5361-z
60. Martin J, Frederick A, Luo Y et al. Generation and characterization of adeno-associated virus producer cell lines for research and preclinical vector production. Hum. Gene Ther. Methods 2013; 24(4): 253–69.
https://doi.org/10.1089/hgtb.2013.046
61. Thorne BA. Towards industrial scale manufacturing of MYDICAR® – An AAV gene therapy product for heart failure. In: Presented at the 5h Spring Meeting of ISBiotech, Washington DC, 9–11 March, 2015.
62. Rice SA, Knipe DM. Genetic evidence for two distinct transactivation functions of the herpes simplex virus alpha protein ICP27. J. Virol. 1990; 64(4): 1704–15.
63. Clément N, Knop DR, Byrne BJ. Large-scale adeno-associated viral vector production using a herpesvirus-based system enables manufacturing for clinical studies. Hum. Gene Ther. 2009; 20(8): 796–806.
https://doi.org/10.1089/hum.2009.094
64. Knop DR, Harrell H. Bioreactor production of recombinant herpes simplex virus vectors. Biotechnol. Prog. 2007; 23(3): 715–21.
https://doi.org/10.1021/bp060373p
65. Kang W, Wang L, Harrell H et al.. An efficient rHSV-based complementation system for the production of multiple rAAV vector serotypes. Gene Ther. 2009; 16(2): 229–39.
https://doi.org/10.1038/gt.2008.158
66. Adamson-Small L, Potter M, Falk DJ et al.. A scalable method for the production of high –titer and high-quality adeno-associated type 9 vectors using the HSV platform. Mol. Ther. Methods Clin. Dev. 2016; 3: 16031.
https://doi.org/10.1038/mtm.2016.31
67. Thomas DL, Wang L, Niamke J et al. Scalable recombinant adeno-associated virus production using recombinant herpes simplex virus type 1 coinfection of suspension-adapted mammalian cells. Hum. Gene Ther. 2009; 20(8): 861–70.
https://doi.org/10.1089/hum.2009.004
68. Knop DR. AAV manufacturing using herpes –assisted vector expansion, In: Presented at the 5th Spring Meeting of ISBiotech, Washington/DC, 9–11 March, 2015.
69. Ye GJ, Scotti MM, Thomas DL et al. Herpes simplex virus clearance during purification of a recombinant adeno-associated virus serotype 1 vector. Hum. Gene Ther. Clin. Dev. 2014; 25(4): 212–7.
https://doi.org/10.1089/humc.2014.060
70. Urabe M, Ding C, Kotin RM. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum. Gene Ther. 2002; 13(16): 1935–43.
https://doi.org/10.1089/10430340260355347
71. Kohlbrenner E, Aslanidi G, Nash K et al. Successful production of pseudotyped rAAV vectors using a modified baculovirus expression system. Mol. Ther. 2005; 12(6): 1217–25.
https://doi.org/10.1016/j.ymthe.2005.08.018
72. Girod A, Wobus CE, Zádori Z et al. The VP1 capsid protein of adeno-associated virus type 2 is carrying a phospholipase A2 domain required for virus infectivity. J. Gen. Virol. 2002; 83(5): 973–8.
https://doi.org/10.1099/0022-1317-83-5-973
73. Smith RH, Levy JR, Kotin RM. A simplified baculovirus-AAV expression vector system coupled with one-step affinity purification yields high-titer rAAV stocks from insect cells. Mol. Ther. 2009; 17(11): 1888–96.
https://doi.org/10.1038/mt.2009.128
74. Cecchini S, Virag T, Kotin RM. Reproducible high yields of recombinant adeno-associated virus produced using invertebrate cells in 0.02- to 200-liter cultures. Hum. Gene Ther. 2011; 22(8): 1021–30.
https://doi.org/10.1089/hum.2010.250
75. Chen H. Intron splicing-mediated expression of AAV Rep and Cap genes and production of AAV vectors in insect cells. Mol. Ther. 2008; 16(5): 924–30.
https://doi.org/10.1038/mt.2008.35
76. Chen H. Adeno-associated virus vectors for human gene therapy. World J. Med. Genet. 2015; 5(3): 28–45.
https://doi.org/10.5496/wjmg.v5.i3.28
77. Hermens WTJMC, Haast SJP, Biesmans DJ et al. AAV vectors with improved rep coding sequences for production in insect cells. WO2007/148971 A2.
78. Urabe M, Ozawa K, Haast SJP et al. Improved AAV vectors produced in insect cells. WO2007/046703 A2.
79. Bédard C, Perret S, Kamen AA. Fed-batch cultures of Sf-9 cells supports 3 x 107 cells per ml and improves baculovirus-expressed recombinant protein yields. Biotechnol. Lett. 1997; 19(7): 629–32.
https://doi.org/10.1023/A:1018378529299
80. Bhatia R, Jesionowski G, Ferrance J et al. Insect cell physiology. Cytotechnology 1997; 24(1): 1–9.
https://doi.org/10.1023/B:CYTO.0000010410.24541.dd
81. Ikonomou L, Schneider YJ, Agathos SN. Insect cell culture for industrial production of recombinant proteins. Appl. Microbiol. Biotechnol. 2003; 62(1): 1–20.
https://doi.org/10.1007/s00253-003-1223-9
82. Palomares LA, Lopez S, Ramirez OT. Utilization of oxygen uptake rate to assess the role of glucose and glutamine in the metabolism of infected insect cell cultures. Biochem. Eng. J. 2004; 19(1): 87–93.
https://doi.org/10.1016/j.bej.2003.12.002
83. Taticek RA, Shuler ML. Effect of elevated oxygen and glutamine levels on foreign protein production at high cell densities using the insect cell baculovirus expression system. Biotechnol. Bioeng. 1997; 54(2): 142–52.
https://doi.org/10.1002/(SICI)1097-0290(19970420)54:23.0.CO;2-L
84. Mena JA, Aucoin MG, Montes J et al. Improving adeno-associated vector yield in high density insect cell cultures. J. Gene Med. 2010; 12(2): 157–67.
https://doi.org/10.1002/jgm.1420
85. Rivollet L, Savy A, Sanatine P et al. Kinetic studies of co-infection of Sf9 cells be recombinant baculoviruses at low MOI in view of rAAV production – process improvement. Presented at the 24th Annual Congress of ESGCT, Florence/I, 18-21 October 2016, poster P399.
86. Dickx Y, Savy A, Nauwynck L et al.. Comparison of truncated and wildtype ITRs: Impact on AAV vector production and quality. Presented at the 16th International Parvovirus Workshop, Ajaccio/France, 19-23 June 2016, poster.
87. Samulski RJ, Srivastava A, Berns KI et al. Rescue of adeno-associated virus from recombinant plasmids: gene correction within the terminal repeats of AAV. Cell 1983; 33(1): 135–43.
https://doi.org/10.1016/0092-8674(83)90342-2
88. Samulski RJ, Chang L-S, Shenk T. A recombinant plasmid from which an infectious adeno-associated virus genoms can be excised in vitro and its use to study viral replication. J. Virol. 1987; 61(10): 3096–101.
89. Wang XS, Ponnazhagan S, Srivastava A. Rescue and replication of adeno-associated virus type 2 as well as vector DNA sequences from recombinant plasmids containing deletions in the viral inverted terminal repeats: selective encapsidation of viral genomes in progeny virions. J. Virol. 1996; 70(3): 1668–77.
90. Noordman Y, Lubelski J, Bakker AC. Mutated rep encoding sequences for use in AAV production. US 2013/0023034.
91. Slack JM, Kuzio J, Faulkner P. Characterization of v-cath, a cathepsin L-like proteinase expressed by the baculovirus Autographa californica multiple nuclear polyhedrosis virus. J. Gen. Virol. 1995; 76(5): 1091–8.
https://doi.org/10.1099/0022-1317-76-5-1091
92. Hawtin RE, Zarkowska T, Arnold K et al. Liquefaction of Autographa californica nucleopolyhedrovirus-infected insects is dependent on the integrity of virus-encoded chitinase and cathepsin genes. Virology 1997; 238(2): 243–53.
https://doi.org/10.1006/viro.1997.8816
93. Galibert L, Merten O-W, van Oers M et al. Improved baculovirus expression systems. WO2013/014294 A2.
94. Cohen DPA, Marek M, Davies BG et al. Encyclopedia of Autographa californica Nucleopolyhedrovirus Genes. Virologica Sinica 2009; 24(5): 359-414
https://doi.org/10.1007/s12250-009-3059-7
95. Merten O-W, Galibert L, Jacob A. Système baculovirus pour l’expression d’un vecteur de thérapie génique. WO 2013/014400 A1.
96. Aslanidi G, Lamb K, Zolotukhin S. An inducible system for highly efficient production of recombinant adeno-associated virus (rAAV) vectors in insect Sf9 cells. Proc. Natl. Acad. Sci. USA 2009; 106(13): 5059–64.
https://doi.org/10.1073/pnas.0810614106
97. Mietzsch M, Casteleyn V, Weger S et al. OneBac 2.0: Sf9 Cell Lines for Production of AAV5 Vectors with Enhanced Infectivity and Minimal Encapsidation of Foreign DNA. Hum. Gene Ther. 2015; 26(10): 688–97.
https://doi.org/10.1089/hum.2015.050
98. Hauck B, Murphy SL, Smith PH, et al. Undetectable transcription of cap in a clinical AAV vector: implications for preformed capsid in immune responses. Mol. Ther. 2009; 17(1): 144–152.
https://doi.org/10.1038/mt.2008.227
99. Mietzsch M, Grasse S, Zurawski C et al. OneBac: platform for scalable and high-titer production of adeno-associated virus serotype 1-12 vectors for gene therapy. Hum. Gene Ther. 2014; 25(3): 212–22.
https://doi.org/10.1089/hum.2013.184
100. Rabinowitz JE, Rolling F, Li C et al. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J. Virol. 2002; 76(2): 791–801.
https://doi.org/10.1128/JVI.76.2.791-801.2002
101. Ling C, Yin Z, Li J et al. Strategies to generate high-titer, high-potency recombinant AAV3 serotype vectors. Mol. Ther. Methods Clin. Dev. 2016; 3: 16029.
https://doi.org/10.1038/mtm.2016.29
102. Hu YC, Kaufman J, Cho MW et al. Production of HIV-1 gp120 in packed-bed bioreactor using the vaccinia virus/T7 expression system. Biotechnol. Prog. 2000; 16(5): 744–50.
https://doi.org/10.1021/bp000112u
103. Ramirez A, Steel M, Jing F et al. A highly scalable baculovirus-based process to manufacture AAV vector of various serotypes. Presented at the 24th Annual Congress of the European Society of Gene & Cell Therapy, Florence, Italy, 18–21 October 2016, P389.
104. Nathwani A. Progress for gene therapy in haemophilia. Presented at the 24th Annual Congress of the European Society of Gene & Cell Therapy, Florence, Italy, 18–21 October, 2016, INV93.
105. Guianvarc’h L, Collaud F, Le Bec C et al. Extensive comparison of HEK293 transfection process and baculovirus expression system to manufacture an AAV8-based treatment for Crigler-Najjar syndrome. Presented at the 23rd Annual Congress of the European Society of Gene & Cell Therapy, Helsinki/FIN, 17–20 September 2016, OR069.
106. Booth MJ, Mistry A, Li X et al. Transfection-free and scalable recombinant AAV vector production using HSV/AAV hybrids. Gene Ther. 2004; 11(10): 829–37.
https://doi.org/10.1038/sj.gt.3302226
107. Park JY, Lim BP, Lee K et al. Scalable production of adeno-associated virus type 2 vectors via suspension transfection. Biotechnol. Bioeng. 2006; 94(3): 416–30.
https://doi.org/10.1002/bit.20776
108. Merten O-W. Introduction to animal cell culture technology – past, present and future. Cytotechnology 2006; 50: 1–7.
https://doi.org/10.1007/s10616-006-9009-4
109. Zhang HS, Kim E, Lee S et al. Transduction of striatum and cortex tissues by adeno-associated viral vectors produced by herpes simplex virus- and baculovirus-based methods. J. Virol. Methods 2012; 179(1): 276–80.
https://doi.org/10.1016/j.jviromet.2011.10.004
110. Green M, Pina M. Biochemical studies on adenovirus multiplication, vi. Properties of highly purified tumorigenic human adenoviruses and their DNA. Proc. Natl Acad. Sci. USA 1964; 51: 1251–9.
https://doi.org/10.1073/pnas.51.6.1251
111. Burova E, Ioffe E. Chromatographic purification of recombinant adenoviral and adeno-associated viral vectors: methods and implications. Gene Ther. 2005; 12(Suppl. 1): S5–17.
https://doi.org/10.1038/sj.gt.3302611
112. Merten O-W, Schweizer M, Chahal P et al. Manufacturing of viral vectors for gene therapy: part II. Downstream processing and safety aspects. Pharm. Bioprocess. 2014; 2: 237–51.
https://doi.org/10.4155/pbp.14.15
113. Sommer JM, Smith PH, Parthasarathy S et al. Quantification of adeno-associated virus particles and empty capsids by optical density measurement. Mol. Ther. 2003; 7(1): 122–8.
https://doi.org/10.1016/S1525-0016(02)00019-9
114. Wright JF, Zelenaia O. Vector characterization methods for quality control testing of recombinant adeno-associated viruses. Methods Mol. Biol. 2011; 737: 247–78.
https://doi.org/10.1007/978-1-61779-095-9_11
115. Manno CS, Pierce GF, Arruda VR et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006; 12(3): 342–7.
https://doi.org/10.1038/nm1358
116. Bennicelli J, Wright JF, Komaromy A et al. Reversal of blindness in animal models of leber congenital amaurosis using optimized AAV2-mediated gene transfer. Mol. Ther. 2008; 16(3): 458–65.
https://doi.org/10.1038/sj.mt.6300389
117. Maguire AM, High KA, Auricchio A et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet 2009; 374(9701): 1597–605.
https://doi.org/10.1016/S0140-6736(09)61836-5
118. Mingozzi F, Anguela XM, Pavani G et al. Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci. Transl. Med. 2013; 5(194): 194ra92.
https://doi.org/10.1126/scitranslmed.3005795
119. Gao K, Li M, Zhong L et al. Empty Virions In AAV8 Vector Preparations Reduce Transduction Efficiency And May Cause Total Viral Particle Dose-Limiting Side-Effects. Mol. Ther. Methods Clin. Dev. 2014; 1(9): 20139.
https://doi.org/10.1038/mtm.2013.9
120. Zolotukhin S, Byrne BJ, Mason E et al. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999; 6(6): 973–85.
https://doi.org/10.1038/sj.gt.3300938
121. Hermens WT, ter Brake O, Dijkhuizen PA et al. Purification of recombinant adeno-associated virus by iodixanol gradient ultracentrifugation allows rapid and reproducible preparation of vector stocks for gene transfer in the nervous system. Hum. Gene Ther. 1999; 10(11): 1885–91.
https://doi.org/10.1089/10430349950017563
122. Qu G, Bahr-Davidson J, Prado J et al. Separation of adeno-associated virus type 2 empty particles from genome containing vectors by anion-exchange column chromatography. J. Virol. Methods 2007; 140(1-2): 183–92.
https://doi.org/10.1016/j.jviromet.2006.11.019
123. Davidoff AM, Ng CY, Sleep S et al. Purification of recombinant adeno-associated virus type 8 vectors by ion exchange chromatography generates clinical grade vector stock. J. Virol. Method 2004; 121(2): 209–15.
https://doi.org/10.1016/j.jviromet.2004.07.001
124. Okada T, Nonaka-Sarukawa M, Uchibori R et al. Scalable purification of adeno-associated virus serotype 1 (AAV1) and AAV8 vectors, using dual ion-exchange adsorptive membranes. Hum. Gene Ther. 2009; 20(9): 1013–21.
https://doi.org/10.1089/hum.2009.006
125. Oranje PPA, Verheesen P, Verbart D et al. Isolation of an adeno-associated virus (AAV)-specific camelid-derived single chain antibody fragment: A novel tool for purification of AAV vectors of different serotypes. Mol. Ther. 2004; 9: S162.
https://doi.org/10.1016/j.ymthe.2004.06.345
126. Hebben M. Debottlenecking downstream process of AAV9 gene therapy vector using customized chromatography resin. Presented at BioInnovation Leaders Summit, London, UK, 11 February, 2015.
127. Potter M, Chesnut K, Muzyczka N et al. Streamlined large-scale production of recombinant adeno-associated virus (rAAV) vectors. Methods Enzymol. 2002; 346: 413–30.
https://doi.org/10.1016/S0076-6879(02)46069-7
128. Wang Q, Lock M, Prongay AJ et al. Identification of an adeno-associated virus binding epitope for AVB sepharose affinity resin. Mol. Ther. Methods Clin. Dev. 2015; 2: 15040.
https://doi.org/10.1038/mtm.2015.40
129. Lecomte E, Tournaire B, Cogné B et al. Advanced Characterization of DNA Molecules in rAAV Vector Preparations by Single-stranded Virus Next-generation Sequencing. Mol. Ther. Nucleic Acids. 2015; 4: e260.
https://doi.org/10.1038/mtna.2015.32
130. Gao G, Wang Q, Wang L et al. Inadvertent gene transfer of co-packaged Rep and Cap sequences during the production of AAV vector and its potential impact on vector performance. Mol. Ther. 2008; 16: S105.
131. Chulay JD, Ye GJ, Thomas DL et al. Preclinical evaluation of a recombinant adeno-associated virus vector expressing human alpha-1 antitrypsin made using a recombinant herpes simplex virus production method. Hum. Gene Ther. 2011; 22(2): 155–65.
https://doi.org/10.1089/hum.2010.118
Affiliations
Otto-Wilhelm Merten
Généthon, Evry, France

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.