Challenges in gene therapy for desminopathies
Cell Gene Therapy Insights 2016;2(4), 403-406.
10.18609/cgti.2016.052



Desminopathies are diseases linked to mutations in the desmin gene. Although of monogenetic etiology, desminopathies are challenging and very intriguing from a gene therapeutic point of view as pathophysiology varies with different mutations. Animal models mirroring their distinct pathophysiologic mechanisms are still scarce. Recent scientific advances have revealed promising new therapeutic strategies. This editorial highlights the recent gene therapeutic approaches being tested in animal models for the treatment of desminopathies.
Desminopathies are diseases linked to mutations in the desmin gene (DES). Clinical manifestations include skeletal muscle weakness, respiratory dysfunction, cardiomyopathy, cardiac conduction disease and, infrequently, changes in bowel habits [1,2]. Most patients exhibit a combined phenotype comprising neurological and cardiologic symptoms, while 22% show an isolated cardiac phenotype [3]. Cardiac involvement has been reported in 74% of patients and is the most life-limiting feature of desminopathy [4]. Variable phenotypes have been described for the same mutations and correlations between mutations and specific symptomatic patterns have been discussed controversially [3]. Disease onset has been described in the first decade for severe autosomal recessive forms and the second to fourth decade for autosomal dominant variants [1].
The first animal models for desminopathies were desmin knockout mice, followed by mice carrying a deletion and mice carrying a point mutation [5–7]. Although of monogenetic etiology, desminopathies pose significant challenges for gene therapeutic approaches owing to inherent pathophysiologic differences. DES codes for a type III intermediate filament protein featuring a typical structure allowing the formation of coiled-coil dimers [8]. These dimers subsequently form filamentous networks that are mainly found in muscle cells. Its critical role in maintaining the cellular structural integrity is evident by its direct physical interactants, including desmoplakin, ankyrin, syncoilin, nebulin, lamin B, myotubularin, myospryn and plectin, linking its filamentous network to the nucleus, the cell membrane, the dystroglycan network, sarcomers, lysosomes and mitochondria (Figure 1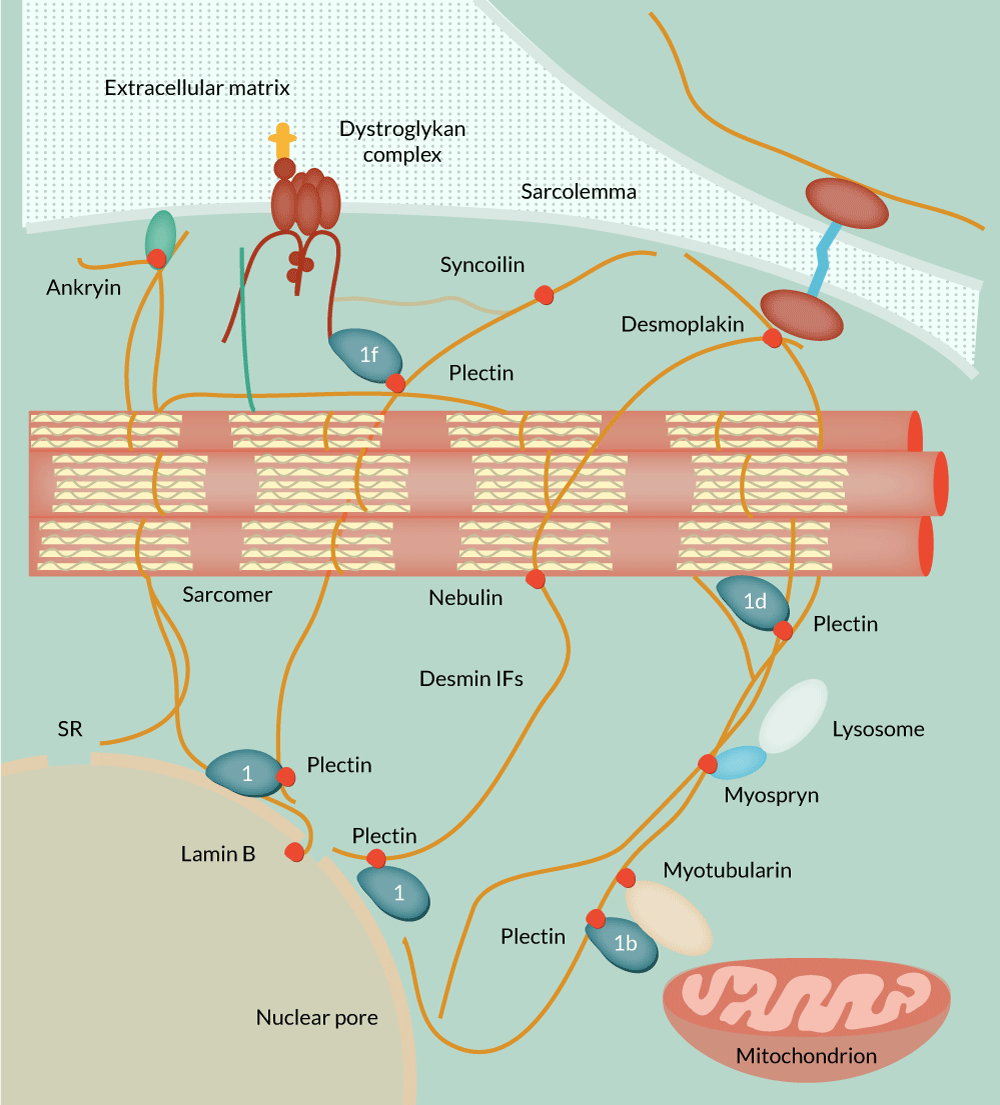
Desmin’s ability to form functional filamentous networks greatly depends on an undisturbed life cycle comprising protein expression, filament formation and protein degradation. DES mutations affect different steps of the protein’s life cycle warranting distinct gene therapeutic approaches. Lack of desmin expression has been reported in humans and successfully treated with recombinant adeno-associated viral (AAV) therapy in mice [9,10]. Recessive mutations might also benefit from AAV-mediated wild-type desmin expression, although no research has been conducted yet, to prove this assumption.
The majority of known DES mutations, however, lead to protein misfolding and are inherited in an autosomal dominant manner [4]. Misfolded desmin exerts a dominant negative effect on wild-type desmin filament assembly and impairs the ubiquitin proteasome system, leading to the formation of desmin rich protein aggregates, which are less likely to be subjected to autophagy [2,11,12]. Although a critical ratio of wild-type to mutant desmin is necessary for mutant desmin to exhibit this effect, a simple increase in wild-type desmin expression might probably just increase protein aggregates [2].
Downregulating mutant desmin expression with selective siRNA might represent a more sophisticated approach. However, engineering siRNAs able to reliably distinguish mRNAs differing only in one base exceeds the method’s potential. A less specific approach using siRNA-mediated downregulation of endogenous desmin expression combined with vector-mediated expression of wild-type desmin might shift the ratio of mutant to wild-type desmin to a sufficient extend in order to attenuate or prevent the dominant negative effect of mutant desmin [2]. This approach would require high levels of recombinant gene expression as endogenous desmin expression makes up 2% of the total protein mass in cardiac tissue [13].
Genome editing might offer a more elegant solution to this problem. An AAV-mediated CRISPR-Cas9 approach has been used in Duchenne muscular dystrophy (DMD) in postnatal and adult mice resulting in 37 and 67% of fibers expressing dystrophin in skeletal muscle, respectively [14–16]. With a relative expression level of 8%, dystrophin expression was still significantly lower in treated DMD animals than in wild-type controls [15]. As the pathophysiology of DMD resembles the type of desminopathy characterized by a lack of desmin expression, results from these studies might not easily transfer to desmin mutations exhibiting a dominant negative effect. However, with further optimization of efficiency, this method might become a promising tool targeting these mutations.
Finally, as changes in protein folding, recycling and degradation are a well-studied pathophysiologic feature accompanying desmin’s life cycle, facilitating the correct function of these processes represents another therapeutic target. Mutations in alpha-B-crystallin, desmin’s main chaperone, show similar cellular and clinical phenotypes as autosomal dominant DES mutations [17]. Overexpression of wild-type alpha-B-crystallin is an interesting approach to decrease protein aggregates and promote proper filament formation. Aside from gene therapy, chemical chaperons, such as 4-phenylbutyrate, have also shown promising results [18]. In conclusion, desminopathies are challenging and very intriguing from a gene therapeutic point of view as pathophysiology varies with different mutations. Animal models mirroring their distinct pathophysiologic mechanisms are still scarce. Recent scientific advances have revealed promising new therapeutic strategies.
financial & competing interests disclosure
The author has no relevant financial involvement with an organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock options or ownership, expert testimony, grants or patents received or pending, or royalties. No writing assistance was utilized in the production of this manuscript.

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.
References
1. Clemen CS, Herrmann H, Strelkov SV, Schröder R. Desminopathies: pathology and mechanisms. Acta Neuropathol. 2013; 125: 47–75. DOI
2. Bär H, Goudeau B, Wälde S et al. Conspicuous involvement of desmin tail mutations in diverse cardiac and skeletal myopathies. Hum. Mutat. 2007; 28: 374–86.
DOI
3. van Spaendonck-Zwarts KY, van Hessem L, Jongbloed JDH et al. Desmin-related myopathy. Clin. Genet. 2011; 80: 354–66. DOI
4. Goldfarb LG, Dalakas MC. Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J. Clin. Invest. 2009; 119: 1806–13. DOI
5. Li Z, Colucci-Guyon E, Pinçon-Raymond M et al. Cardiovascular lesions and skeletal myopathy in mice lacking desmin. Dev. Biol. 1996; 175: 362–6. DOI
6. Wang X, Osinska H, Dorn GW et al. Mouse model of desmin-related cardiomyopathy. Circulation 2001; 103: 2402–7. DOI
7. Clemen CS, Stöckigt F, Strucksberg K-H et al. The toxic effect of R350P mutant desmin in striated muscle of man and mouse. Acta Neuropathol. 2015; 129: 297–315. DOI
8. Fuchs E, Weber K. Intermediate filaments: structure, dynamics, function, and disease. Annu. Rev. Biochem. 1994; 63: 345–82. DOI
9. Heckmann MB, Bauer R, Jungmann A et al. AAV9-mediated gene transfer of desmin ameliorates cardiomyopathy in desmin-deficient mice. Gene Ther. 2016; 23: 673–9. DOI
10. Carmignac V, Sharma S, Arbogast S et al. G.O.7 A homozygous desmin deletion causes an Emery-Dreifuss like recessive myopathy with desmin depletion. Neuromuscul. Disord. 2009; 19: 600. DOI
11. Liu J, Chen Q, Huang W et al. Impairment of the ubiquitin-proteasome system in desminopathy mouse hearts. FASEB J. 2006; 20: 362–4. DOI
12. Wong ESP, Tan JMM, Soong W-E et al. Autophagy-mediated clearance of aggresomes is not a universal phenomenon. Hum. Mol. Genet. 2008; 17: 2570–82. DOI
13. Price MG. Molecular analysis of intermediate filament cytoskeleton–a putative load-bearing structure. Am. J. Physiol. 1984; 246: H566-72. Website
14. Tabebordbar M, Zhu K, Cheng JKW et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016; 351: 407–11. DOI
15. Nelson CE, Hakim CH, Ousterout DG et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016; 351: 403–7. DOI
16. Long C, Amoasii L, Mireault AA et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016; 351: 400–3. DOI
17. McLendon PM, Robbins J. Desmin-related cardiomyopathy: an unfolding story. Am. J. Physiol. Heart Circ. Physiol. 2011; 301: H1220–8. DOI
18. Winter L, Staszewska I, Mihailovska E et al. Chemical chaperone ameliorates pathological protein aggregation in plectin-deficient muscle. J. Clin. Invest. 2014; 124: 1144–57. DOI
19. Heckmann MB. Development of a Gene Therapeutic Approach to Cardiomyopathy in Desmin-Deficient Mice. Heidelberg University, 2014.
Affiliations
Markus B Heckmann, Hugo A Katus and Oliver J Müller
Internal Medicine III, University Hospital Heidelberg, Im Neuenheimer Feld 410, 69120 Heidelberg, Germany
DZHK (German Center for Cardiovascular Research), partner site Heidelberg/Mannheim, Germany