
Manufacturing process development of ATMPs within a regulatory framework for EU clinical trial & marketing authorisation applications
Cell Gene Therapy Insights 2016; 2(4), 425-452.
10.18609/cgti.2016.056
The quality attributes of advanced therapy medicinal products (ATMPs) that correlate with safety and efficacy in patients are determined not only by manufacturing process inputs such as starting and raw materials, but also by how the manufacturing process itself is designed and controlled. To ensure regulatory compliance, the manufacturing process should therefore be developed based on thorough characterization of the ATMP during all stages of process and analytical development; this ensures that the critical quality attributes that correlate with safety and efficacy are identified and that their specifications can be met during routine manufacturing. In the European Union, the regulatory approval of ATMPs for use in patients requires that data demonstrating their quality, safety and efficacy are submitted in dossiers to regulatory agencies for review. Indeed, such dossiers have a specific format that, in the case of quality data in particular, is informative for the manufacturing process development strategy. This manuscript describes how dossier requirements can be implemented into the design of industrialized ATMP manufacturing processes and fulfilled to enable effective regulatory submissions.
The pace of cell and gene therapy development has increased tremendously in recent years, and the technologies for their manufacture have improved to support robust, efficient and cost-effective production at industrial scale [1]. Based on the design of the manufacturing process and/or the therapeutic use of the final product, most cell and gene therapies are now classified as medicinal products in many global territories [2]. This classification as medicinal products (drugs) therefore means that these therapies must comply with stringent regulatory requirements if they are to be authorized for use in patients, either as investigational medicinal products (IMPs) in clinical trials or as licensed medicinal products for sale on international pharmaceutical markets. This is exemplified in the European Union (EU) where medicinal products comprising human cells, genes or tissues are regulated as advanced therapy medicinal products (ATMPs) to distinguish their use from medical procedures involving cell or tissue transplantation.
ATMPs were introduced as a new class of medicinal product in the EU through the so-called ATMP Regulation [3], which came into effect in late 2008 following the recognition by the European Medicines Agency (EMA; the agency responsible for the scientific evaluation, supervision and safety monitoring of medicines developed by pharmaceutical companies for use in the EU) [4] that cell-, gene- and tissue-based therapies needed more rigorous evaluation than was allowed for by medicinal product legislation in force previously. Products containing cells or tissues are defined as ATMPs if they are manufactured using a process that involves substantial manipulation of the starting materials (cells or tissues), while cells or tissues used in medical procedures only undergo processing via minimal manipulation (and are therefore not considered starting materials for product manufacture). Minimal manipulation may simply involve cell purification (without culture) and/or washing before infusion into a patient (hematopoietic stem cell transplantation being the classic example), and so any processing that is more complex would be considered substantial manipulation (e.g., extended culture and passaging to expand a cell population that will be used as a cell therapy). Gene therapies comprise a separate category of ATMPs whose mechanism of action involves the expression of transgenes. They are manufactured through processes that involve the generation of genetic constructs and their amplification in cell lines, following which they are either purified for direct administration (in vivo gene therapies), or used for transduction of therapeutic cells (ex vivo gene therapies). Both cell therapies and gene therapies are considered to work through a pharmaceutical, metabolic or immunological mode of action [3], another aspect of their classification as medicinal products. Human or animal cells and tissues can also undergo substantial manipulations to manufacture products for the repair or regeneration of damaged or diseased tissues and organs, generally in combination with a scaffold (so-called tissue-engineered products). However, such products would be classified as gene therapies if the component cells were genetically modified. Therefore, ATMPs (somatic cell therapy medicinal products [SCTMPs], gene therapy medicinal products [GTMPs] and tissue-engineered products [TEPs], as defined [3]) are regulated as medicinal products because their mode of action is typical of other medicinal products, and/or their production involves substantial manipulation and industrial manufacturing processes. The exception to this rule is when cells are only manipulated minimally but are used for a purpose not reflecting the same essential function of the cells in the recipient as in the donor; in this case, such non-homologous therapeutic use of cells means that they are regulated as a medicinal product, i.e., an ATMP.
To determine whether a therapeutic product based on human cells, genes or tissues meets the criteria that define ATMPs, developers can apply for an ATMP Classification [5,6] from the EMA’s Committee for Advanced Therapies, the expert body responsible for reviewing the quality, safety and efficacy of ATMPs [4].
The aim of medicinal product development is to demonstrate quality, safety & efficacy
The real implication of the classification of cell and gene therapies as medicinal products under the ATMP Regulation is that their development should address the requirements of a medicinal product to successfully obtain, via non-clinical studies and clinical trials, a license for commercial use, i.e., a Marketing Authorisation (MA) [4]. Despite the fact that certain schemes (not discussed here) exist that enable patients to be treated with unlicensed cell or gene therapies, the requirements for achieving MA should not be underestimated, even for autologous products manufactured on a single batch per-patient basis or orphan medicinal products for the treatment of rare diseases. Such products are typical of the eight ATMPs that have already received a MA (Table 1). To receive a Clinical Trial Authorisation (CTA) or a MA, the quality, safety and efficacy of ATMPs must be demonstrated. During product development (Figure 1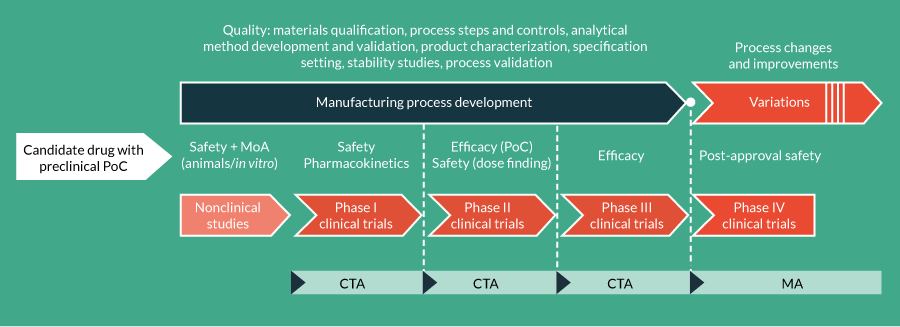
| Table 1: ATMPs to have received an EU Marketing Authorization. | ||||
|---|---|---|---|---|
| Common name(MAH) | Drug substance | Drug product | ATMP class | Current approval status |
| Zalmoxis (MolMed S.p.A.) | Allogeneic T cells genetically modified with a retroviral vector encoding for a truncated form of the human low affinity nerve growth factor receptor (ΔLNGFR) and the herpes simplex I virus thymidine kinase (HSV-TK Mut2) | Zalmoxis 5-20 x 106 cells/mL dispersion for infusion | SCTMP (ex-vivo) | Conditional MA (2016) Orphan designation |
| Strimvelis (GlaxoSmithKline Trading Services Limited) | Autologous CD34+ enriched cell fraction that contains CD34+ cells transduced with retroviral vector that encodes for the human adenosine deaminase (ADA) cDNA sequence from human hematopoietic stem/progenitor (CD34+) cells | Strimvelis 1-10 x 106 CD34+ cells/mL | GTMP (ex-vivo) | Standard MA (2016) |
| Imlygic (Amgen Europe B.V.) | Talimogene laherparepvec | Imlygic 106 plaque forming units /mL solution for injection Imlygic 108 plaque forming units/mL solution for injection | GTMP (in vivo) | Standard MA (2015) |
| Holoclar (Chiesi Farmaceutici S.p.A.) | Ex vivo-expanded autologous human corneal epithelial cells containing stem cells | 7.9 x 104–3.16 x 105 cells/cm2 living tissue equivalent | TEP | Conditional MA (2015) Orphan designation |
| Provenge (Dendreon UK Ltd) | Autologous peripheral blood mononuclear cells including a minimum of 50 million autologous CD54+ cells activated with prostatic acid phosphatase granulocyte-macrophage colony-stimulating factor | 5 x 107 CD54 + cells/250 mL dispersion for infusion | SCTMP | Withdrawn |
| MACI (Vericel Denmark ApS) | Matrix-applied characterized autologous cultured chondrocytes | 0.5–1 x106 /cm2 implantation matrix | TEP | Suspended |
| Glybera (uniQure Biopharma B.V.) | Alipogene tiparvovec | 1 mL of solution, containing 3 × 1012 genome copies | GTMP (in vivo) | MA under Exceptional Circumstances (2012) |
| ChondroCelect (TiGenix N.V.) | Characterized viable autologous cartilage cells expanded ex vivo expressing specific marker proteins | 4 x 106 autologous human cartilage cells in 0.4 mL cell suspension, corresponding to a concentration of 10,000 cells/µL | TEP | Withdrawn |
| To date, eight ATMPs have received a Marketing Authorisation (MA) from the European Commission, although two have been withdrawn by the Marketing Authorisation Holders and one has been suspended by the EMA. Note that different types of MA have been granted to the various ATMPs: Conditional MAs and MAs under Exceptional Circumstances are granted when clinical data supporting the MAA are not extensive, e.g., in the case of medicinal products for rare diseases that qualify for orphan designation. The information presented in this table is taken from the EMA website and is current at the time of going to press. | ||||
The data from the product development phase are presented in a dossier submitted to regulatory agencies for evaluation ahead of CTA or MA. For a CTA application, quality, safety and efficacy data are submitted in an Investigational Medicinal Product Dossier (IMPD) [7], while a Marketing Authorisation Application (MAA) follows more strictly the Common Technical Document (CTD) format [8] in which quality data are presented in Module 3, and safety and efficacy data in Modules 4 and 5 (nonclinical and clinical study reports, respectively). However, for quality data in particular, IMPD Section 2 (where the quality data are reported) follows essentially the same structure as CTD Module 3. The structure of CTD Module 3 is presented in Table 2; it is composed of three main sections on Drug Substance, Drug Product (both of which are defined later) and Appendices, each with subsections on specific aspects of quality development. In effect, the structure of CTD Module 3 therefore provides a plan for how a quality development strategy should be designed and executed.
| Outline structure of Module 3 of the Common Technical Document. | |
|---|---|
| Section | Subsection |
| 3.2.S DRUG SUBSTANCE | |
| 3.2.S.1 General Information | 3.2.S.1.1 Nomenclature |
| 3.2.S.1.2 Structure | |
| 3.2.S.1.3 General Properties | |
| 3.2.S.2 Manufacture | 3.2.S.2.1 Manufacturer(s) |
| 3.2.S.2.2 Description of Manufacturing Process and Process Controls | |
| 3.2.S.2.3 Control of Materials | |
| 3.2.S.2.4 Controls of Critical Steps and Intermediates | |
| 3.2.S.2.5 Process Validation and/or Evaluation | |
| 3.2.S.2.6 Manufacturing Process Development | |
| 3.2.S.3 Characterization | 3.2.S.3.1 Elucidation of Structure and other Characteristics |
| 3.2.S.3.2 Impurities | |
| 3.2.S.4 Control of Drug Substance | 3.2.S.4.1 Specification |
| 3.2.S.4.2 Analytical Procedures | |
| 3.2.S.4.3 Validation of Analytical Procedures | |
| 3.2.S.4.4 Batch Analyses | |
| 3.2.S.4.5 Justification of Specifications | |
| 3.2.S.5 Reference Standards or Materials | |
| 3.2.S.6 Container Closure System | |
| 3.2.S.7 Stability | 3.2.S.7.1 Stability Summary and Conclusions |
| 3.2.S.7.2 Post-approval Stability Protocol and Stability Commitment | |
| 3.2.S.7.3 Stability Data | |
| 3.2.P Drug Product | |
| 3.2.P.2 Pharmaceutical Development | 3.2.P.1 Description and Composition of the Drug Product |
| 3.2.P.2.2 DRUG PRODUCT | |
| 3.2.P.2.3 Manufacturing Process Development | |
| 3.2.P.2.4 Container Closure System | |
| 3.2.P.2.5 Microbiological Attributes | |
| 3.2.P.2.6 Compatibility | |
| 3.2.P.3 Manufacture | 3.2.P.3.1 Manufacturer(s) |
| 3.2.P.3.2 Batch Formula | |
| 3.2.P.3.3 Description of Manufacturing Process and Process Controls | |
| 3.2.P.3.4 Controls of Critical Steps and Intermediates | |
| 3.2.P.3.5 Process Validation and/or Evaluation | |
| 3.2.P.4 Control of Excipients | 3.2.P.4.1 Specifications |
| 3.2.P.4.2 Analytical Procedures | |
| 3.2.P.4.3 Validation of Analytical Procedures | |
| 3.2.P.4.4 Justification of Specifications | |
| 3.2.P.4.5 Excipients of Human or Animal Origin | |
| 3.2.P.4.6 Novel Excipients | |
| 3.2.P.5 Control of Drug Product | 3.2.P.5.1 Specification(s) |
| 3.2.P.5.2 Analytical Procedures | |
| 3.2.P.5.3 Validation of Analytical Procedures | |
| 3.2.P.5.4 Batch Analyses | |
| 3.2.P.5.5 Characterisation of Impurities | |
| 3.2.P.5.6 Justification of Specification(s) | |
| 3.2.P.6 Reference Standards or Materials | |
| 3.2.P.7 Container Closure System | |
| 3.2.P.8 Stability | 3.2.P.8.1 Stability Summary and Conclusion |
| 3.2.P.8.2 Post-approval Stability Protocol and Stability Commitment | |
| 3.2.P.8.3 Stability Data | |
| 3.2.A APPENDICES | |
| Section | |
| 3.2.A.1 Facilities and Equipment | |
| 3.2.A.2 Adventitious Agents Safety Evaluation | |
| 3.2.A.3 Excipients | |
| The Common Technical Document (CTD) describes the format for MAA submissions in the EU. Module 3 is used to present CMC data generated during the quality development stage, and the format of Module 3 is also used in Section 2 of the IMPD used to support CTA applications. Module 3 is divided into three sections on drug substance, drug product and appendices, and the content required in each section is shown here. A medicinal product quality development strategy therefore requires a full understanding and characterization of the manufacturing processes for both drug substance and drug product. In combination with appropriate guidance documents (see text for details), Module 3 can be considered to provide a blueprint for developing ATMP quality through CMC. Guidance on the CTD itself can be found in [8], and on the IMPD in [7]. | |
Guidance on developing quality, safety & efficacy: importance to ATMP developers
Although the legal framework regulating ATMPs goes far beyond the ATMP Regulation alone [Manuscript in Preparation], this regulation is the starting point for determining (as discussed above) whether a cell or gene therapy will be regulated as a medicinal product and will therefore need to meet the legal requirements of quality, safety and efficacy for patient use. The legal framework for all medicinal products does not, however, describe how quality, safety and efficacy should be demonstrated, because each development programme is product-specific. Rather, guidance documents are the instruments used to implement the specific aspects of quality, safety and efficacy on a product-by-product basis. Various guidance documents are available from different sources, many of which are applicable, either directly or indirectly, to ATMPs. In the EU, the key sources of guidance documents for ATMP developers are the EMA’s Scientific Guidelines [9], the International Council for Harmonisation (ICH) guidelines [10] and the European Pharmacopeia [11]. The importance to ATMP developers of following the appropriate guidelines cannot be understated if the aim is to achieve CTA or MA. Two guidelines in particular should be considered essential for developers of ATMPs, these being the Guideline on human cell-based medicinal products (which is applicable to SCTMPs and TEPs) [12] and the Guideline on the quality, non-clinical and clinical aspects of gene therapy medicinal products (which is applicable to both in vivo and ex vivo GTMPs) [13]. For the purposes of this discussion, these and other quality development guidelines will be identified as appropriate throughout the text but it will not be possible to elaborate fully on their content. Note that in cases where cells and tissues of animal origin are used in the development of an ATMP, additional guidance [14] should also be consulted.
ATMP quality is developed through a thorough understanding of the manufacturing process, its inputs & its outputs: the role of CMC
The aim of regulatory submissions (CTAs and MAAs) is to demonstrate the quality, safety and efficacy of a medicinal product, and this requires thorough knowledge of the product’s characteristics and the processes used to manufacture it in addition to nonclinical and clinical data [4]. A medicinal product manufacturing process can be considered, in regulatory terms, to be comprised of two manufacturing processes: manufacture of the drug substance (DS); and manufacture of the drug product (DP). The DS may also be called the ‘active substance’ or ‘active pharmaceutical ingredient’, because it is the component of the medicinal product that is responsible for its pharmaceutical activity. For the different classes of ATMPs, the DS is the manipulated cells for SCTMPs, the gene-modified cells for ex vivo GTMPs, the nucleic acid sequence or modified microorganism/virus for in vivo GTMPs, and the manipulated cells and/or tissues for TEPs [3,15]. The DS is manufactured from starting materials and raw materials: the starting materials for ATMPs would be tissues, cells and/or genetic vector stocks while the raw materials may include, for example, culture medium, serum, growth factors, enzymes and chemical reagents such as detergents and saline buffers. The DP can be considered to be the DS in its final formulation for administration to patients. As such, the DS may be blended with excipients (non-pharmaceutical ingredients that are essential to the DP composition; e.g., a cryopreservation medium for SCTMPs, or a stabilizing agent for in vivo GTMPs) and will be filled into an appropriate container for storage and shipping (see Table 1 for descriptions of the DS and DP of ATMPs that have received a MA). All aspects of medicinal product manufacturing process development must therefore address starting materials, raw materials, excipients, the DS, the DP and the DS and DP containers in a way that is appropriate for regulatory review ahead of CTA or MA. Together, the studies performed to demonstrate medicinal product quality through manufacturing process development in this way are referred to as CMC (chemistry, manufacturing and controls).
As the term suggests, CMC has its roots in the development of small molecule drugs. By extrapolation to ATMPs, the chemistry element can be considered to mean the raw materials, starting materials and excipients used, and also the composition of the process outputs (intermediates, DS and DP); the manufacturing element can be considered to mean how the manufacturing process(es) is (are) developed and validated; and the controls element can be considered to mean how the manufacturing processes are shown to operate within parameters such that their outputs (DS and DP) meet the specifications shown to correlate with safety and efficacy in clinical use. Therefore, a manufacturing process that is controlled on all levels (inputs, process steps, analytical methods, outputs, validation) is key to the development of medicinal product quality, and should be presented accordingly, in CTD Module 3 format, for the review of EU regulatory submissions.
Mapping a developmental manufacturing process to CTD Module 3
The structure of CTD Module 3 (Table 2) [8] can be thought of as providing a blueprint for developing a CMC strategy to create the final, controlled ATMP manufacturing process. As a first step in mapping a developmental manufacturing process to CTD Module 3, the starting materials, the DS and the DP should be defined, thereby splitting the overall process into sub-processes as they will be presented in Module 3 and enabling each to be addressed specifically and appropriately. Definition of starting materials, DS and DP can be aided by the EMA’s ATMP Classification procedure [4], and starting this procedure early in development can therefore be advantageous to the implementation of an effective CMC strategy. Once the starting materials, DS and DP have been defined, a process for manufacturing an ATMP from the starting materials through the DS and DP stages can be outlined, as shown schematically in Figure 2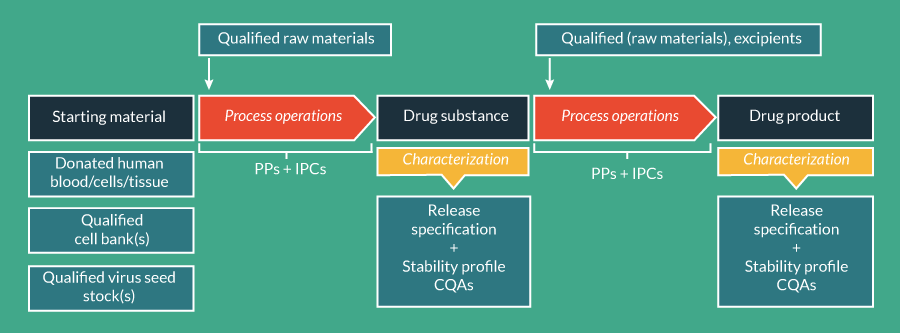
The ultimate goal of CMC is a controlled manufacturing process
Understanding how the combination of all operations involved contributes to the composition and activity of the final product (in terms of both DS and DP) is highly important when developing an ATMP manufacturing process [16]. In reality, manufacturing processes for ATMPs are much more complex than outlined in Figure 2, involving multiples steps and possibly intermediates. Controlling all aspects of the process is therefore crucial – and essential at the point of critical steps and intermediates – to ensure that the desired output (i.e., a product with defined CQAs linked to nonclinical and clinical data) is obtained. Process control is achieved through monitoring of defined process parameters (e.g., bioreactor settings such as temperature, agitation speed, feed rate) and in-process controls (IPCs) for which acceptance criteria are set. The purpose of IPCs is to enable continuous monitoring of the quality attributes of DS and DP such that potential issues that may lead to an out-of-specification product can be identified. Therefore, all process steps and intermediate products that contribute to a product’s quality attributes, including the critical steps and intermediates that determine CQAs, should be monitored through IPCs. Examples of how IPCs could be used for ATMPs may include the monitoring of cell confluence, cell morphology, cell population doubling level (PDL) and doubling time (Td), cell yield and viability on passaging, viral genome copy number and bioburden of intermediate products. However, specific IPCs should be assigned on a product-by-product basis, the full complement of which will require that the DS and DP manufacturing processes are fully mapped out in terms of their component steps. Acceptance criteria should be set and justified based on data obtained during the development phase that demonstrate robustness of the process in terms of generating DS and DP with the requisite quality attributes, which are characterized as discussed below.
Extensive product characterization is needed for the development of product quality through specification setting
Control of DS & DP through critical quality attributes
As discussed earlier, the concept of medicinal product quality can be considered to be the development of a specification defined by CQAs that correlate with safe and efficacious clinical use. Scientific Guidelines on ATMP development published by the EMA [12,13] describe which product characteristics should be used as CQAs for SCTMPs, GTMPs and TEPs, and it is important to note that specifications apply to both the DS and DP components of these products. In principle, the basis for DS release testing is to demonstrate that it is of the appropriate quality for use in DP manufacture, while the basis for DP release testing is to demonstrate that the final product is of the appropriate quality for use in patients. However, it will be seen later that sometimes it is necessary to release final products on the basis of a DS specification, or that DS specifications should instead be applied to an intermediate as a surrogate, thus further highlighting the importance of fully characterizing both DS and DP.
There are differences between those CQAs required for cell-based ATMPs and in vivo GTMPS, but potency, content, identity, purity and impurities are of primary importance for all ATMPs and should be characterized using product-specific assays (referred to as analytical methods in the CTD). Other CQAs are somewhat generic but are considered essential for patient safety, for example, microbiological sterility, absence of mycoplasma and absence of endotoxins (tests that, for all ATMPs, should be performed according to validated standard European Pharmacopoeia methods, unless otherwise justified in the regulatory submission).
Specifications for sterility and mycoplasma are absolute values (sterile, not detected) but other specifications (potency, content, impurities) will be reported as a range of values based on data from batches manufactured during development. These data should be used to set upper and lower limits dictated by manufacturing process experience from batches shown to be safe and effective in nonclinical and clinical studies, or from batches used for process validation and verification.
Potency is a quantitative measure of how a medicinal product’s biological activity correlates with or predicts the desired therapeutic effect. Potency is unique in being the only CQA linked directly to efficacy, rather than product composition. Various biological activities considered appropriate to a product’s mechanism of action in patients should be assessed during development to determine, through analysis of clinical data, which most reliably indicates a product’s potency for the release specification [4,12,13,17,18]. Depending on whether an ATMP is considered to work through a pharmaceutical, metabolic or immunological mode of action, or be involved in tissue repair and regeneration, potency assays may be based on expression of markers of specific cell types whose content is linked to efficacy; expression or secretion of specific molecules and analysis of their biological activity; infectivity of a viral vector (together with biological activity of the expressed product); or in vitro lysis of target cells (e.g., tumor cells). Surrogate assays may sometimes need to be developed to obtain timely results when mechanisms of action are complex.
Identity assays for cell-based ATMPs will be based on phenotypic and/or genotypic analysis of the expression of specific markers for one or more cell populations. Where the DS is a heterogeneous cell population, the identity markers may also be used to quantify the content of each population, and the content data may then be used to determine the purity of the different cell types. For in vivo GTMPs, identity assays are needed to demonstrate the nucleotide sequences of the vector and therapeutic gene, as well as the protein composition of capsids in the case of a viral vector. Purity of a viral vector may be measured as a percentage of the total viral particles that contain a virus genome or, alternatively, the proportion of viral particles that are able to productively infect permissive cells.
Impurities may be either product- or process-related. Product-related impurities for cell-based ATMPs may include non-viable cells, cells from tissue biopsies that do not contribute to the therapeutic mechanism, or a progenitor cell type when the DS is defined by a differentiated cell population. Product-related impurities for in vivo GTMPs may include residual therapeutic protein expressed by the vector and empty virus particles. At least some product-related impurities will form part of the DS and DP specifications. Process-related impurities will be derived from the raw materials used and must be controlled because, depending on the nature of the raw material, residual amounts in the final product may have toxic or immunogenic activities in patients. Therefore, the manufacturing process will need to be validated to show that these materials are cleared from the final product. Contamination with adventitious agents represents another potential process-related impurity. Although starting materials and raw materials should be controlled for adventitious agents prior to use, they may still be introduced during the process or amplified in culture even when tests (which are based on limit of detection) provide negative results. Consideration should therefore be given to the use of in-process controls and the testing of intermediate products where appropriate, even though appropriate facilities controls should also be in place to prevent contamination.
Specifications for CQAs such as potency, identity, purity and impurities can only be developed by thorough characterization to determine, for the purpose of selecting potency and identity assays, which biological activities and phenotypic properties of the product may best predict clinical benefit; and, for the purpose of selecting assays for content, purity and impurities, how the process contributes to the overall composition of the DS and DP. Ultimately, all specifications must be justified based on the analysis of data from manufactured batches, and so it is important to implement as many assays as considered appropriate for full characterization starting in early development such that sufficient data are available to make an informed decision on which assays should be chosen for CQAs – and subsequently fully validated – in late development (assays should be suitably qualified in early development to show that they are fit for purpose and generate reliable data). A scientific and technical justification must be provided in the MAA in case certain release tests cannot be performed. In such circumstances, adequate in-process controls should be incorporated into the manufacturing process, supported by the results of the clinical studies. In addition to the ATMP-specific guidelines [8,12], the ICH Q6B guideline Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products [17] also provides guidance of relevance to ATMP developers on product characterization and setting of specifications. Further characterization is needed to determine other quality aspects of ATMPs that will not become part of the DS or DP specification but are predictive of safety and efficacy, for example tumorigenicity potential and karyotypic stability for SCTMPs and TEPs (the primary guidelines on ATMPs provide a full discussion [12,13]).
Demonstrating the stability of DS & DP
Stability testing is required to generate data in support of shelf lives of DS, DP (in the final container) and any intermediates for presentation in the IMPD and MAA, and these data should be acquired throughout the product development phase as part of the characterization studies. However, DS stability testing is not always relevant when the DS is immediately processed into the DP. Stability testing should incorporate a similar analytical testing plan as is used for product characterization, although it may be necessary to use stability-specific tests either additionally or instead to create a stability-indicating profile. While not all characterization and stability assays will be available and/or fully validated until the later stages of development, it is important to begin a stability testing program with available assays as early as possible in development because stability data need to be included in regulatory submissions throughout the clinical development phase even prior to MAA. Guidance on stability studies can be found in the ATMP-specific scientific guidelines [12,13] and also in Note for Guidance on Quality of Biotechnological Products: Stability Testing of Biotechnological/Biological Products CPMP/ICH/138/95 [19]. Like the ICH Q6B guideline [17], this latter guideline was written for biological/biotechnological products but is acknowledged as also having relevance for ATMPs. For ATMPs, in-use stability should also be studied to determine the time window within which the product should be used at the point of use. Various scenarios can be envisioned, which for cell-based ATMPs primarily concern their viability as an indicator of stability. For example, the in-use stability of TEPs, which because of their nature are likely to be shipped as non-cryopreserved products immediately on DP formulation, may only be a few hours and will also include the shipping time until the product reaches the surgical site. Similarly, cells that cannot be cryopreserved at the DP stage will need to be shown to withstand shipping times and conditions as part of their in-use stability profile. Considerations for products that can be cryopreserved at the DP stage will need to include how they are further stored at point of use and also the timeframe in which they are used on controlled thawing, which may require time for further preparation (such as compounding of lots) as well as time for administration to the patient.
Container closure systems
Another aspect of overall product characterization and stability testing is the determination of the suitability of container closures systems for DS and/or DP, data for which are presented in specific sections of CTD Module 3. Even for ATMPs that have a short in-use shelf life, a container closure system of some description (e.g., transfusion bags or cryovials) in which the final product is formulated and transported from the manufacturing site to the point of care will be needed. For any product formulation, the container closure system must be demonstrated to be suitable. This may include suitability in terms of its potential effects on the CQAs of the product, compatibility with the product in terms of any extractable or leachable components that may contaminate the product, compatibility with the formulation in terms of adsorption of the active substance and/or excipients to surfaces, compatibility with storage temperature (including freeze–thaw manipulations for cryopreserved products) and suitability in terms of maintaining a product’s quality during transport via the means that will be used to distribute the clinical or commercial product. Appropriate evaluation or validation studies will therefore need to be undertaken.
Manufacturing process development & comparability studies
Regulatory agencies understand that manufacturing processes will evolve during the developmental phase to improve the product through the refinement of specifications and/or the use of more suitable materials, implementation of more efficient/robust process steps and controls, or use of improved analytical methods. Indeed, the role of process development experimentation is to create a fixed process comprising defined operations, PPs, IPCs and DS/DP/intermediate quality attributes such that those critical to determining product quality can be identified. Changes in manufacturing processes made for these purposes must therefore be demonstrated to maintain or improve product quality (in terms of characterization data and process controls), and this is the basis of comparability studies. Comparability studies therefore compare the quality of a medicinal product manufactured according to the current and proposed new processes using some or all of the assays implemented to characterize the product, control the process, develop the release specifications and determine stability. Comparability studies are typically performed at the level of product quality initially, and clinical data will only be required if comparability cannot be demonstrated through CMC and/or nonclinical studies. Guidance on comparability studies that can be applied to ATMPs is provided in the ICH Q5E guide¬line, Comparability of Biotechnological/Biological Products Subject to Changes in their Manufacturing Process [20].
Technology transfer & process validation
The final step in manufacturing process development of medicinal products is process validation, which is done in parallel to or following the successful pivotal clinical trial to ensure that the commercial process being proposed in the MAA is fit for purpose (i.e., can reproducibly deliver a product with the requisite quality attributes predictive of safety and efficacy at the scale required to supply the market). At this stage, all analytical methods should also be validated according to the ICH Q2B guideline Validation of Analytical Procedures: Methodology [21]. Prior to clinical trials earlier in development, process validation in the strict sense does not apply. Rather, process robustness is shown through technology transfer of the process from the development laboratory to the manufacturing facility [22]. Technology transfer of a process will typically involve, first, shakedown runs to adapt the process to GMP requirements and finalize operating procedures; second, engineering runs to confirm the procedures and demonstrate robustness and reproducibility of the process in support of initiation of clinical manufacturing; and third, GMP manufacturing runs to supply product to clinical trials. DS, DP and intermediates from engineering and GMP runs are typically used in characterization and stability studies.
Validation of the commercial process is carried out ideally at industrial scale as required for commercial supply. Historically, a minimum of three successful engineering runs has been considered necessary to demonstrate that the process is validated, but product requirements should be evaluated individually through a risk-based approach that takes into account process and product knowledge together with continuous validation requirements over the product lifecycle.
Controlling ATMP quality through process inputs: starting materials, raw materials & excipients
Starting materials
An ATMP’s ultimate specification is a reflection of data generated during development to characterize its full complement of quality attributes and identify those that are critical to safety and efficacy. Quality attributes are essentially intrinsic properties of the starting materials used in product manufacture, and the quality of the starting material therefore influences the quality of the product. For a cell-based ATMP, for example, this may be in terms of the ability to obtain the cell population with the desired therapeutic activity in sufficient quantity and/or eliminate undesirable cellular impurities or contamination with adventitious agents that may overtake the culture or put patient safety at risk.
Starting materials for ATMP manufacture are cells, vector stocks or tissues of human and/or animal origin, and this presents the need to control the spread of disease-causing and other adventitious agents (viruses, bacteria, fungi, mycoplasma and transmissible spongiform encephalopathy [TSE] agents). Cells may be either autologous or allogeneic donations (e.g., a leukapheresis sample), or a qualified, GMP-grade cell bank (e.g., pluripotent stem cells or a cell line used to amplify a viral vector). Vector stocks (e.g., adenovirus or retrovirus constructs containing therapeutic gene sequences and any associated helper viruses) can be considered analogous to cell banks, and therefore should also be appropriately qualified and manufactured in accordance with GMP. Finally, tissues may be either autologous or allogeneic donations (e.g., a corneal biopsy). The source and nature of the starting material will therefore determine the appropriate control measures for its use in ATMP development and manufacture.
Donated cell and tissue starting materials should be procured in accordance with the donor consent and screening procedures described in the EU Tissues and Cells Directives [23–26] or the Blood Directive plus its three technical directives [27–30] to prevent the transmission of diseases. Additionally for such starting materials, it will likely be necessary to set, as appropriate, further quality parameters with associated acceptance criteria, including bioburden, absence of mycoplasma, delivery time from the procurement site, tissue size/volume/physical form, total cell number obtained and viability of the cells obtained. Donated tissues may often be non-sterile when procured and so initial processing steps may need to be performed in the presence of antibiotics to reduce the bioburden. However, antibiotics will need to be removed from the process prior to DP formulation and be demonstrated to not present a process-related impurity.
Cell banks and virus seed stocks are required to be manufactured in accordance with certain guidelines to ensure their suitability for ATMP manufacture. Specifically, cell bank starting materials should be manufactured and characterized in accordance with the ICH Q5D guideline, Derivation and Characterization of Cell Substrates Used for Production of Biotechnological/Biological Products [31]. The typical approach described in this guideline is to create a two-tiered cell bank, in which a master cell bank (MCB) is initially produced from which one or more working cell banks (WCBs) are then derived to guarantee that the capacity of the bank is sufficient to enable continued supply of the starting material (i.e., the WCB) for commercial manufacture without depleting the MCB. This is of both financial and operational importance because the level of characterization and testing required for the MCB is greater than for the WCB. Therefore, if a fully characterized MCB can be shown to be stable over a long period of time, it is quicker and cheaper to derive new WCBs from it when required rather than derive one or more MCBs, which would also need to be demonstrated to be suitable for medicinal product manufacture through comparability studies. Characterization of cell banks according to ICH Q5D is carried out to ensure, among other things, sterility, absence of adventitious agents (also taking into account the ICH Q5A guideline [32]), cell identity, purity, karyotypic stability and MCB/WCB stability. Note that qualification of cell bank starting materials in this way (which is usually done by a contract manufacturer) does not reduce or negate the need to characterize drug substances and drug products derived from them, which should always be done according to the appropriate guidelines [12,13] as discussed above.
Starting materials for GTMPs will depend on the vector being used. As an example, those for adenovirus-associated (AAV)-based vectors such as Glybera (Table 1) [33] will include the virus seed stock (or stocks when helper vectors are needed to assemble the complete therapeutic virus particle) and the producer cell line for amplifying and packaging the virus [34]. In common with cell lines, virus seed stocks should be banked as master and working virus seeds (MSVs and WSVs) whenever possible. The producer cell line(s) will need to be qualified according to ICH Q5D [31] as described above, while the MSV and WSV are characterized as described in the GTMPs guideline [13]. Characterization should include the confirmation of genetic (genome sequence) and phenotypic (capsid/envelope proteins) identity, virus concentration and infectious titer, genome integrity, expression of the therapeutic sequence, phenotypic characteristics, biological activity of therapeutic sequence, absence of replication competent virus, inter-vial homogeneity, sterility and absence of mycoplasma. Additionally, the complete sequence of the therapeutic and regulatory elements, and where feasible the complete sequence of the virus in the seed bank, should be confirmed.
Raw materials & excipients
As well as starting materials, the potential for contamination with disease-transmitting and other adventitious agents is also a risk for raw materials or excipients of biological origin, both human- and animal-derived (e.g., bovine serum, porcine trypsin and human fibrin). The selection of such materials should therefore be carried out in consideration of European Pharmacopoeia monographs and specific EMA Scientific Guidelines [35,36], including those for human donors [24–30], to ensure that viral and TSE risks are eliminated. Risks posed by xenogeneic viruses and TSE agents (particularly bovine spongiform encephalopathy) mean that animal-derived materials should be avoided if possible, and full qualification based on risk assessment is necessary when they must be used.
Other than bovine serum, water for injections and miscellaneous chemical reagents, very few pharmacopeial-grade raw materials suitable for use in the manufacture of cell- and gene-based medicinal products are currently available, and out of necessity, non-pharmacopeial grade (research-grade) materials must be used instead. For pharmacopeial-grade materials, no further testing is required if appropriate certification for suitability and microbial tests can be provided by the supplier. If it is not possible to use pharmacopeial-grade materials, the developer should implement tests (either in-house or through a contract testing laboratory) for assessed risks and provide justified acceptance criteria for the materials selected. This is true for raw materials of both biological and non-biological origin.
Risks associated with non-biological raw materials and excipients may include contamination with bacteria, fungi and/or bacterially derived endotoxins, lack of a desired activity, presence of undesirable impurities, or absence of data confirming the identity of the material itself. Assessing the risks may also require that suppliers are audited to ensure the suitability of their own facilities, processes and materials, and service level agreements should be in place to ensure the quality of the materials supplied. This level of qualification also applies to materials that may be labeled as GMP-grade, which essentially is an indicator that a material has been manufactured in accordance with a GMP quality management system in a licensed facility and should not be interpreted as an indicator of suitability for use in medicinal product manufacture. Volume 9 of the European Pharmacopoeia, due to come into effect on 01 January 2017, will contain a new general chapter on “Raw materials of biological origin for the production of cell-based and gene therapy medicinal products for human use” that will provide guidance on how certain non-pharmacopeial raw materials used in ATMP manufacture should be selected and qualified. The guidance provided will be similar to that in Chapter 1043 of the US Pharmacopeia [37], which is applicable for cell and gene therapies being developed in the USA but may also be useful in principle to EU developers.
Sterility is a crucial element of the specification for ATMPs. Therefore, risks to sterility presented by raw materials, many of which may be combined into a culture medium for cell growth, must be controlled. Culture media are typically sterile filtered prior to use, but filtration alone is not sufficient to guarantee sterility because the efficiency of a sterilizing filter depends on the bioburden level in the material to be filtered. Therefore, the bioburden level in all non-sterile components of a culture medium should be determined (according to pharmacopeial methods) prior to filtration to ensure that the bioburden level in the complete medium is not too high to be cleared by filtration. Applicable guidance is provided in an EMA Scientific Guideline on finished dosage forms [38]. Furthermore, the efficiency of sterile filtration should be tested using pharmacopeial sterility or filter-integrity tests. Certain excipients that may be used in the formulation of ATMPs will also have pharmacopeial monographs; they can therefore be considered suitable in terms of their quality but should also be characterized with respect to their combination with the active substance. For excipients used for the first time in ATMP development, the regulatory requirements for novel excipients in Annex I of Directive 2001/83/EC1 [39] apply.
Specific CMC considerations for common AMTP manufacturing scenarios
To illustrate how the principles discussed above apply to real-world ATMP development programs [40], considerations for manufacturing ATMPs from donated tissue or banked cell starting materials will be outlined. These considerations will also need to take into account whether the manufacturing process is based on a scale-out (small batch size; e.g., autologous products, one batch per patient) or scale-up (large batch size; e.g., allogeneic products, multiple identical doses per batch) approach, and the specific bioprocessing technologies involved [1]. The development of many ATMPs actually begins from donated tissue starting materials, and mesenchymal stem cells (MSCs) will be used to highlight principles that will also apply to other types of ATMPs, such as ex vivo GTMPs and TEPs. Banked cell starting materials will instead be discussed from the perspective of in vivo GTMPs.
Manufacturing ATMPs from donated tissue starting materials
MSCs are commonly used adult stem cells for ATMP development [41] that can be isolated from donated tissue starting materials such as bone marrow, umbilical cord blood and adipose tissue [42], which are typically of allogeneic origin although autologous tissues may also be used [43]. However, in common with other cells isolated from donated tissues, their bioprocessing at industrial scale presents a number of challenges [44,45] and concerns related to the heterogeneity and donor-to-donor variability of the starting materials from which they are obtained, the low percentage of MSCs within these starting materials, the finite lifetime of the cells in culture and the development of appropriate potency assays reflective of clinical mode of action [46–51].
Various bioprocessing technology options may be employed for large-scale manufacturing of MSCs once isolated from the starting material, but in all cases the manufacturing process is based on expansion of a selected cell population expressing the MSC phenotype [52]. It is this expansion stage that determines how the characterization and control strategy should be designed and implemented in consideration of the chosen bioprocessing technology. The objective of the expansion phase is to increase the yield of MSCs with the desired phenotype, i.e., to obtain sufficient MSCs for therapeutic use within their finite in vitro lifespan [53,54]. This latter characteristic of MSCs, together with potentially low initial yields from some tissue biopsies [55], means that one biopsy can only be used to manufacture a limited number of batches of the final MSC-based product. The process must therefore be designed to be sufficiently robust and reproducible to manufacture multiple batches of an ATMP with consistent specifications from any number of biopsies of the appropriate tissue type. Process robustness and reproducibility are ensured through IPCs applied to different stages of the manufacturing process, such as the monitoring of PDL, cell Td, morphology and confluence, the timing of cell detachment steps, and the control of any cell washing, concentration and filling procedures.
The simplest manufacturing process scenario would involve continuous expansion of MSCs isolated from the starting material until sufficient numbers can be harvested to formulate the product. This scenario reflects the basic outline shown in Figure 2; the DS would be the cells at the end of the final passage, and DP manufacture would involve harvesting the cells (DS) from the culture vessels, resuspending them in an appropriate medium and filling them into bags or vials at a defined concentration and volume, ready for dosing. In practice, however, such a simplistic manufacturing process cannot fulfill the requirements of regulatory-compliance for two main reasons.
One problem associated with running the manufacturing process continuously is that the expansion phase may generate cell numbers that cannot be handled efficiently at the formulation phase. Therefore, following an initial expansion phase, it may be appropriate to introduce a cryopreservation step to provide individual cell aliquots for further processing. MSC isolation and expansion will represent an intermediate stage between the starting material and the DS (Figure 3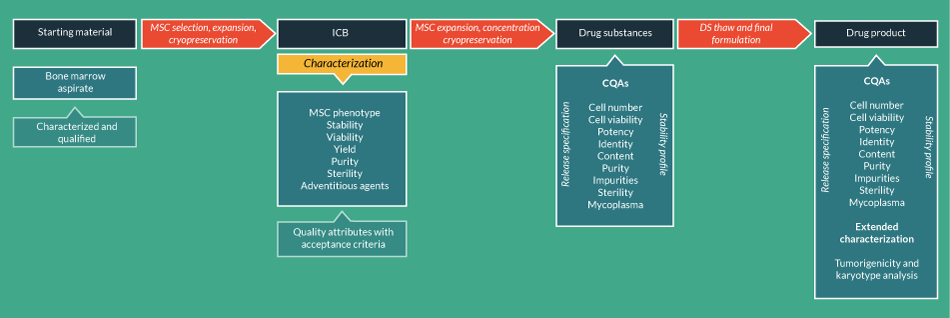
Of greater concern, a second problem associated with running the manufacturing process continuously is that it does not provide the opportunity to obtain release test data confirming the specifications of DS and DP before the final product is used to treat patients. Downstream of ICB production, the next stage in the manufacturing process would be to further expand a number of cells from the ICB until the required number for dosing is obtained within the PDL necessary to deliver the requisite CQAs (potency, identity, viability, purity). At the end of this expansion phase the cells would be cryopreserved again as the DS batch, thus enabling the DS release specifications to be determined using a representative sample of cryopreserved aliquots thawed from the total batch. In this respect, it is possible to determine (in combination with stability studies) that cells thawed from the final cell bank (FCB) comprising the DS will retain the requisite CQAs for use in patients. However, because any further processing of the cells on thawing is likely to be minimal (e.g., washing or dilution), whether the FCB really represents the DP as well as the DS can be questioned. What is important in this situation is that the full complement of CQAs is determined on the FCB and that any additional tests necessary on thawing (e.g., determination of cell numbers and viability) are performed before administration. Agreement with regulatory agencies through scientific advice procedures may be necessary to obtain endorsement for the characterization and control strategy (including definition of DS and/or DP) proposed.
Cryopreservation of the FCB also provides convenience in enabling the cells to be thawed when needed for administration to patients. Ideally, this is carried out at the point of care because the shelf life of the cells will be short. Therefore, the need to ship fresh cells requires careful consideration of end-to-end manufacturing and supply chain logistics [1,56,57]. If developmental studies show that the FCB can be cryopreserved and administered on thawing without further culture, cryopreservation of the FCB provides convenience to the supply chain in being able to manufacture a product that can be shipped frozen and further stored at clinical sites within the shelf life demonstrated by stability studies. In this case, the clinical site will need to be provided with validated instructions on how to prepare the product for administration (e.g., wash to remove cryopreservative).
It is true, however, that not all ATMPs can be cryopreserved to enable release testing to be performed before patient treatment, with TEPs providing the best example. For such products, the DP may need to be released on the basis of DS testing, but DP release tests should still be performed so that specification data can be obtained post-administration for information that may be used to evolve the manufacturing process or inform the results of patient follow-up. For some ATMPs, it may also be appropriate to perform release tests on an ICB as a surrogate for the DS, which may also substitute for DP release testing prior to administration. This reduced testing at the DP stage should be justified based on the relative risks posed by process operations from DS to DP, and appropriate control through the use of carefully selected IPCs should be demonstrated where necessary. The manufacturing process for Holoclar (Table 1) provides a good example [58].
Manufacturing ATMPs using established cell banks as starting materials
Established cell banks that can be used as starting materials for ATMP manufacture may include induced pluripotent stem cells (iPSCs) for the derivation of terminally differentiated cells that may have therapeutic applications [59–62], and producer cell lines used in the production of viral vectors [63,64]. All such cell lines should be manufactured and characterized in accordance with the ICH Q5D guideline [31] and other relevant guidelines [12,13] to qualify them for use as starting materials in ATMP manufacture. Note that cell lines can also be classified as raw materials, for example xenogeneic feeder cells on which human stem cells may be cultured [58].
It is possible that these cell line starting materials could be used to manufacture the DS either indirectly (i.e., via intermediates) or directly. For example, the derivation of terminally differentiated cells (such as mesenchymoangioblast-derived MSCs [65] and retinal pigment epithelial cells [66] as a DS from iPSCs may occur through a direct process, but the production of a viral vector would require several intermediates (Figure 4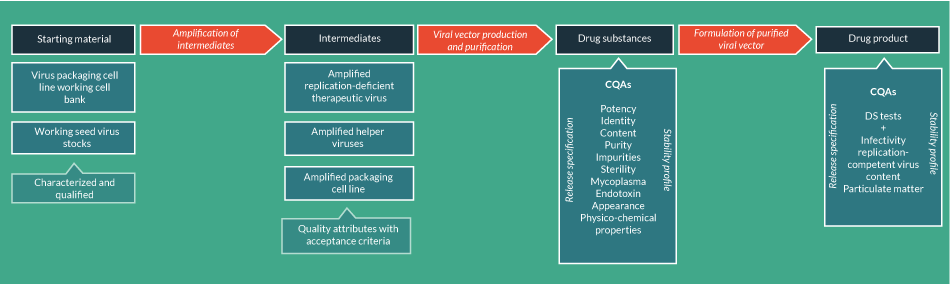
The production of viral vectors involves the generation of a number of other intermediates. In fact, the production of viral vectors is more similar to the production of therapeutic recombinant proteins than cell-based ATMPs, involving expression of the vector in the producer cell line followed by chromatographic purification together with filtration, concentration and buffer exchange. These steps will generate a number of intermediate products that should be subject to IPCs to control the CQAs of the final product. Of course, considerations for DS and DP testing as discussed above for donated tissue-derived ATMPs may also apply to banked cell-derived ATMPs and should be incorporated into the development strategy.
For ATMPs derived from both donated tissue and banked cell starting materials, it is therefore important to determine the approach most appropriate to manufacturing process development and control for any individual product, and this may involve discussions with regulatory agencies at scientific advice meetings.
Conclusions
This article has discussed those aspects of ATMP quality development that should be addressed to enable dossiers suitable for EU regulatory submissions to be compiled. ATMPs comprise a diverse class of products, and although each product will have its own specific quality development requirements, common CMC principles as highlighted here apply to all. Combined ATMPs (incorporating a medical device) [3] create further product diversity but have not been discussed here. However, the same principles described in this manuscript would apply to the ATMP component, although they would additionally need to be thoroughly characterized in combination with the device.
Like all medicinal products in development, not every investigational ATMP will progress successfully through clinical trials to MAA stage, but designing the development program with a MAA in mind can maximize the chances of success for all products and be further enabling for those products that will hit this milestone. For an effective quality development program, understanding the CMC requirements of CTD Module 3 is useful in this respect to enable a manufacturing process to be designed and implemented in line with the common CMC elements that need to be addressed in a regulatory submission, and to keep pace with a successful clinical development programme heading towards pivotal trials and MAA. Product-specific CMC considerations will vary according to nature of the starting material (autologous vs allogeneic), the ATMP classification (SCTMP, GTMP or TEP) and the manufacturing process modality (scale out or scale up; intermediate steps or continuous processing), all of which will impact on how the DS and DP manufacturing processes are designed and controlled to enable appropriate characterisation studies to be performed that will be supportive of product release to patients.
Financial & competing interests disclosure
Anthony Lodge is an employee of Chiesi Farmaceutici S.p.A., which is the marketing authorisation holder of Holoclar. Giulia Detela is an employee of Cell and Gene Therapy Catapult. Cell & Gene Therapy Catapult is an Innovate UK programme. No writing assistance was utilized in the production of this manuscript.
References
1. Mount NM, Ward SJ, Kefalas P et al. Cell-based therapy technology classifications and translational challenges. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2015; 370(1680): 20150017. CrossRef
2. Lodge A, Detela G, Barry J et al. Chapter 10: Global Regulatory Perspective for MSCs. Viswanthan S, Hematti P (Eds). Mesenchymal Stromal Cells, Translational Pathways to Clinical Adoption, Elsevier Inc. (2017).
3. The European Parliament and the Council of the European Union. REGULATION (EC) No 1394/2007 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 13 November 2007 on advanced therapy medicinal products and amending Directive 2001/83/EC and Regulation (EC) No 726/2004. 2007.
4. Salmikangas P, Menezes-Ferreira M, Reischl I et al. Manufacturing, characterization and control of cell-based medicinal products: challenging paradigms toward commercial use. Regen. Med. 2015; 10: 65–78. CrossRef
5. Cuende N, Rico L, Herrera C. Concise Review: Bone Marrow Mononuclear Cells for the Treatment of Ischemic Syndromes: Medicinal Product or Cell Transplantation? Stem Cells Transl. Med. 2012; 1: 403–8. CrossRef
6. Izeta A, Herrera C, Mata R et al. Cell-based product classification procedure: What can be done differently to improve decisions on borderline products? Cytotherapy 2016; 18: 809–15. CrossRef
7. European Medicines Agency. CHMP/QWP/185401/2004 – Guideline on the requirements to the chemical and pharmaceutical quality documentation concerning investigational medicinal products in clinical trials. 2006.
8. European Medicines Agency. CPMP/ICH/2887/99 – Quality – Common Technical Document for the registration of pharmaceuticals for human use. Quality overall summary of Module 2 and Module 3: Quality. 2003.
9. EMA Guidelines
10. ICH Guidelines
11. Council of Europe
12. European Medicines Agency. EMEA/CHMP/410869/2006 – Guideline on human cell-based medicinal products. 2007.
13. European Medicines Agency. EMA/CAT/GTWP/671639/2008 – Guideline on quality, non-clinical and clinical aspects of medicinal products containing genetically modified cells. 2012.
14. European Medicines Agency. EMEA/CHMP/CPWP/83508/2009 – Guideline on Xenogeneic Cell-Based Medicinal Products. 2009.
15. The European Parliament and the Council of the European Union. COMMISSION DIRECTIVE 2009/120/EC of 14 September 2009 amending Directive 2001/83/EC of the European Parliament and of the Council on the Community code relating to medicinal products for human use as regards advanced therapy medicinal products. 2009.
16. Lipsitz YY, Timmins NE, Zandstra PW. Quality cell therapy manufacturing by design. Nat Biotechnol. 2016; 34: 393–400. CrossRef
17. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products Q6B. 1999.
18. European Medicines Agency. EMEA/CHMP/BWP/271475/2006 – Guideline on potency testing of cell based immunotherapiey medicinal products for the treatment of cancer. 2007.
19. European Medicines Agency. CPMP/ICH/138/95 – ICH Topic Q 5 C: Stability Testing of Biotechnological/Biological Products – Step 5 Note for Guidance on Quality of Biotechnological Products: Stability Testing of Biotechnological/Biological Products. 1996.
20. International Council for Harmonisation. Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Proces Q5E. ICH Harmon. Tripart. Guidel. 2004.
21. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Validation of Analytical Procedures: Text and Methodology Q2(R1). 2005.
22. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Pharmaceutical quality system Q10. 2008.
23. The European Commission. DIRECTIVE 2004/23/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 31 March 2004 on setting standards of quality and safety for the donation, procurement, testing, processing, preservation, storage and distribution of human tissues and cells. Off. J. Eur. Union. 2004.
24. The European Commission. COMMISSION DIRECTIVE 2006/17/EC of 8 February 2006 implementing Directive 2004/23/EC of the European Parliament and of the Council as regards certain technical requirements for the donation, procurement and testing of human tissues and cells. 2006.
25. The European Commission. COMMISSION DIRECTIVE 2006/86/EC of 24 October 2006 implementing Directive 2004/23/EC of the European Parliament and of the Council as regards traceability requirements, notification of serious adverse reactions and events and certain technical requirement. 2006.
26. The European Parliament and the Council of the European Union. COMMISSION DIRECTIVE 2012/39/EU of 26 November 2012 amending Directive 2006/17/EC as regards certain technical requirements for the testing of human tissues and cells. 2012.
27. The European Parliament and the Council of the European Union. DIRECTIVE 2002/98/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 27 January 2003 setting standards of quality and safety for the collection, testing, processing, storage and distribution of human blood and blood componentsand amending Directive 2001/. 2006.
28. The European Parliament and the Council of the European Union. COMMISSION DIRECTIVE 2004/33/EC of 22 March 2004 implementing Directive 2002/98/EC of the European Parliament and of the Council as regards certain technical requirements for blood and blood components. 2004.
29. The European Parliament and the Council of the European Union. COMMISSION DIRECTIVE 2005/61/EC of 30 September 2005 implementing Directive 2002/98/EC of the European Parliament and of the Council as regards traceability requirements and notification of serious adverse reactions and events. 2005.
30. The European Parliament and the Council of the European Union. COMMISSION DIRECTIVE 2005/62/EC of 30 September 2005 implementing Directive 2002/98/EC of the European Parliament and of the Council as regards Community standards and specifications relating to a quality system for blood establishments. 2005.
31. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Derivation and Characterisation of Cell Substrates Used for Production of Biotechnological/Biological Products Q5D. 1997.
32. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Viral Safety Evaluation of Biotechnology Products Derived From Cell Lines of Human or Animal Origin Q5A(R1). 1999.
33. Carpentier AC, Frisch F, Labbe SM, et al. Effect of Alipogene Tiparvovec (AAV1-LPLs447x) on Postprandial Chylomicron Metabolism in Lipoprotein Lipase-Deficient Patients. J. Clin. Endocrinol. Metab. 2012; 97: 1635–44. CrossRef
34. Ayuso E. Manufacturing of recombinant adeno-associated viral vectors: new technologies are welcome. Mol. Ther. Methods Clin. Dev. 2016; 3: 15049. CrossRef
35. European Medicines Agency. EMA/CHMP/BWP/814397/2011 – Guideline on the Use of porcine trypsin used in the manufacture of human biological medicinal products. 2013.
36. European Medicines Agency. EMA/CHMP/BWP/303353/2010 – CHMP position statement on Creutzfeldt-Jakob disease and plasma-derived and urine-derived medicinal products CHMP position statement on Creutzfeldt-Jakob disease and plasma-derived and urine-derived medicinal products. 2011.
37. US Pharmacopeia
38. European Medicines Agency. EMA/CHMP/QWP/245074/2015 – Guideline on manufacture of the finished dosage form. 2015.
39. The European Parliament and the Council of the European Union. DIRECTIVE 2001/83/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 6 November 2001 on the Community code relating to medicinal products for human use. 2000.
40. Hanna E, Rémuzat C, Auquier P et al. Advanced therapy medicinal products: current and future perspectives. J. Mark. Access Heal. Policy 2016; 4: 1–10. CrossRef
41. Bisson I, Green E, Sharpe M. Landscape of current and emerging cell therapy clinical trials in the UK: current status, comparison to global trends and future perspectives. Regen. Med. 2015; 10: 169–79. CrossRef
42. Augello A, Kurth TB, de Bari C. Mesenchymal stem cells: A perspective from in vitro cultures to in vivo migration and niches. Eur. Cells Mater. 2010; 20: 121–33. Website
43. Squillaro T, Peluso G, Galderisi U. Clinical Trials with Mesenchymal Stem Cells: An Update. Cell Transplant. 2016; 25: 2015–6. CrossRef
44. Ma T, Tsai AC, Liu Y. Biomanufacturing of human mesenchymal stem cells in cell therapy: Influence of microenvironment on scalable expansion in bioreactors. Biochem. Eng. J. 2016; 108: 44–50. CrossRef
45. Heathman TRJ, Nienow AW, McCall MJ et al. The Translation of Cell-Based Therapies: Clinical Landscape and Manufacturing Challenges. Regen. Med. 2015; 10: 49–64. CrossRef
46. Martin I, De Boer J, Sensebe L. A relativity concept in mesenchymal stromal cell manufacturing. Cytotherapy 2016; 18: 613–20. CrossRef
47. Panchalingam KM, Jung S, Rosenberg L et al. Bioprocessing strategies for the large-scale production of human mesenchymal stem cells: a review. Stem Cell Res. Ther. 2015; 6: 225. CrossRef
48. Wuchter P, Bieback K, Schrezenmeier H et al. Standardization of Good Manufacturing Practice-compliant production of bone marrow-derived human mesenchymal stromal cells for immunotherapeutic applications. Cytotherapy 2015; 17: 128–39.
https:/doi.org/10.1016/j.jcyt.2014.04.002
49. Sensebé L, Bourin P, Tarte K. Good manufacturing practices production of mesenchymal stem/stromal cells. Hum. Gene Ther. 2011; 22: 19–26. CrossRef
50. Galipeau J, Krampera M. The challenge of defining mesenchymal stromal cell potency assays and their potential use as release criteria. Cytotherapy 2015; 17: 125–7. CrossRef
51. Galipeau J, Krampera M, Barrett J et al. Potency Release Criterion for Advanced Phase Clinical Trials. Cytotherapy 2016; 18: 151–9. CrossRef
52. Dominici M, Le Blanc K, Mueller I et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006; 8: 315–7. CrossRef
53. Banfi A, Bianchi G, Notaro R et al. Replicative aging and gene expression in long-term cultures of human bone marrow stromal cells. Tissue Eng. 2002; 8: 901–10. CrossRef
54. Kundrotas G, Gasperskaja E, Slapsyte G et al. Identity, proliferation capacity, genomic stability and novel senescence markers of mesenchymal stem cells isolated from low volume of human bone marrow. Oncotarget. 2016; 7. CrossRef
55. Pittenger MF. Multilineage Potential of Adult Human Mesenchymal Stem Cells. Science 1999; 284: 143–7. CrossRef
56. Trainor N, Pietak A, Smith T. Rethinking clinical delivery of adult stem cell therapies. Nat. Biotechnol. 2014; 32: 729–35. CrossRef
57. Veronesi E, Murgia A, Casselli A et al. Transportation Conditions for Prompt Use of Ex-vivo Expanded and Freshly Harvested Clinical Grade Bone Marrow MSC For Bone Regeneration. Tissue Eng. Part C. Methods 2013; 20: 1–41. CrossRef
58. Milazzo G, De Luca M, Pellegrini G. Holoclar: first of its kind in more ways than one. Cell Gene Ther. Insights 2016; 2: 183–97. CrossRef
59. Barry J, Hyllner J, Stacey G et al. Setting Up a Haplobank: Issues and Solutions. Curr. Stem Cell Reports 2015; 1: 110–7. CrossRef
60. Andrews P, Stacey G, Baker D et al. Points to consider in the development of seed stocks of pluripotent stem cells for clinical applications: International Stem Cell Banking Initiative (ISCBI). Regen. Med. 2015; 10: 1–44. CrossRef
61. Stacey GN, Crook JM, Hei D et al. Banking human induced pluripotent stem cells: Lessons learned from embryonic stem cells? Cell Stem Cell. 2013; 13: 385–8. CrossRef
62. Baghbaderani BA, Tian X, Neo BH et al. CGMP-manufactured human induced pluripotent stem cells are available for pre-clinical and clinical applications. Stem Cell Reports. 2015; 5: 647–59. CrossRef
63. Guenther C, Hauser A, Huss R. Advances in Pharmaceutical Cell Therapy. Principles of cell-based biopharmaceuticals – biology, quality, manufacturing, clinical implementation and economics. World Scientific Publishing Co. Pte. Ltd; 2016.
64. Broussau S, Jabbour N, Lachapelle G et al. Inducible packaging cells for large-scale production of lentiviral vectors in serum-free suspension culture. Mol. Ther. 2008; 16: 500–7. CrossRef
65. Koch JM, Souza SSD, Schwahn DJ et al. Mesenchymoangioblast-derived mesenchymal stromal cells inhibit cell damage , tissue damage and improve peripheral blood flow following hindlimb ischemic injury in mice. Cytotherapy 2016; 18: 219–28. CrossRef
66. Chen FK, McLenachan S, Edel M et al. iPS Cells for Modelling and Treatment of Retinal Diseases. J. Clin. Med. 2014; 3(4): 1511–41. CrossRef
67. Lawler SE, Chiocca EA, Hospital W. Oncolytic Virus-Mediated Immunotherapy: A Combinatorial Approach for Cancer Treatment. J. Clin. Oncol.2016; 33: 2812–4. CrossRef
68. Clément N, Grieger JC. Manufacturing of recombinant adeno-associated viral vectors for clinical trials. Mol. Ther. Methods Clin. Dev. 2016; 3: 16002. CrossRef

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.
Affiliations
Giulia Detela1 and Anthony Lodge2*
1 Cell and Gene Therapy Catapult, 12th Floor Tower Wing, Guy’s Hospital, London, UK
2 Chiesi Farmaceutici S.p.A., Via Palermo 26/A, 43122 Parma, Italy
a.lodge@chiesi.com
*Author for correspondence