
Unlocking the therapeutic and commercial potential of CAR-T technology
Cell Gene Therapy Insights 2016; 2(3), 357-376.
10.18609/cgti.2016.043
The striking clinical results of CAR-T therapies in blood cancers have shifted the cell therapy field from one that has future potential, to one offering hope of a future to those living with cancer. Since 2012 the field has expanded beyond investigators to investment and initial public offerings, individual ‘cures’ and industry growth. Here, we review the current state of CAR-T therapeutics, from improvements in CAR-T technology to clinical results, business growth and future applications. Finally, we discuss the remaining barriers to successful commercialization of CAR-T technology identified by key investors within this rapidly expanding field.
The body’s adaptive immune system provides a powerful and safe way to eliminate health threats. Beginning with vaccines in the late 1700s and moving to the use of antisera, monoclonal antibodies and most recently T cells, researchers have sought to harness people’s own immune systems to fight their own diseases. Today, technology has enabled the genetic modification of T cells to express a chimeric antigen receptor (CAR) that allows them to recognize antigens on malignant cells. This modification eliminates the need for other components of the immune system to present the antigen to the T cell. In turn, this streamlines the process of triggering the cytolytic signaling cascade, making these T cells cancer-killing machines.
In the past few years, CAR-T cells have proven to be a highly effective therapeutic tool in treating certain types of cancers, particularly hematological malignancies. The clinical successes achieved by CAR-T therapies have stimulated interest and participation from industry players. Millions of dollars from private, public and government sectors have been invested in the last few years and collaborations among bio-ventures and academic facilities have been initiated. The increased activity has translated to the optimization of CAR-T constructs at the bench, and the development of applications both within and beyond hematological cancers at the bedside. However, more improvements are required to increase the safety and efficacy of these therapeutics and the advance towards their commercialization. In this article, we review the scientific and clinical evolution of CAR-T cells, their translation into treatments for patients and the commercialization barriers that need to be overcome to unlock the potential of CAR-T therapies.
The business of CAR-T therapeutics
The development of CAR-T cells by Zelig Eshhar and Steven Rosenberg in 1990 was an attempt to improve our ability to use the body’s innate immune system to target and ‘cure’ cancer [1]. More than two decades later, this approach has reached a tipping point. The striking clinical success achieved in treating acute lymphoblastic leukemia (ALL) in the case of Emily Whitehead, the first pediatric patient to be treated with CAR-T therapy in spring 2012, was a game changer. It was this clinical result and the duration of its response that engaged the interest of investors, who have contributed $600 million in venture capital (VC) to directly support the development of CAR-T therapeutics through August 2016 (Figure 1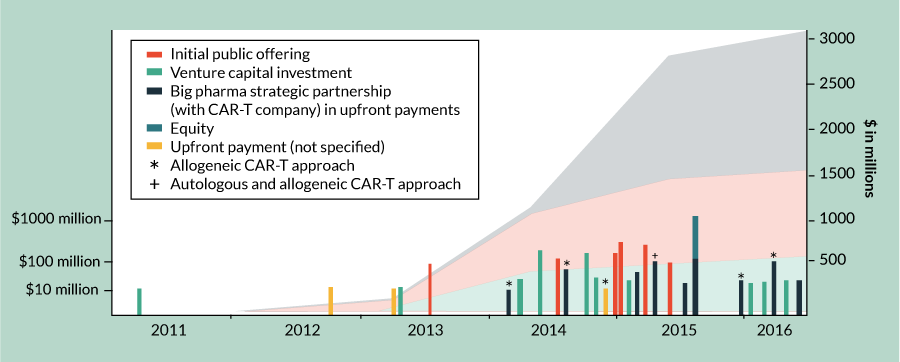
| Table 1 Venture capital investment in CAR-T companies 2011–2016. | |||
| Company | Venture capital ($ in millions) | Date | CAR-Tapproach |
| Kite Pharma | 15 | Mar 2011 | Autologous |
| Kite Pharma | 20 | May 2013 | Autologous |
| Kite Pharma | 50 | Apr 2014 | Autologous |
| Juno | 176 | Apr 2014 | Autologous |
| Juno | 134 | Aug 2014 | Autologous |
| Bellicum | 55 | Aug 2014 | Autologous |
| Autolus | 45 | Jan 2015 | Autologous |
| Poseida | 23 | Dec 2015 | Allogeneic |
| CARsgen | 30 | Jan 2016 | Autologous |
| Autolus | 57 | Mar 2016 | Autologous |
| Total | 605 | ||
Includes only venture capital funding for companies involved in CAR-T program(s) at the time of investment. For example, venture capital funding of Bluebird Bio occurred prior to their CAR-T programs, while the company had only a gene therapy focus. These investments are not included. Source: Company press releases. | |||
Big Pharma’s hesitation to engage in the cell therapy industry also shifted in 2012, with the clinical results of CAR-T in ALL and chronic lymphocytic leukemia (CLL). That year marked the first commercial partnership in this space, between Novartis and the University of Pennsylvania, USA (Figure 1 & Table 3) [3]. Since 2012, seven other Big Pharma companies have placed bets in the CAR-T space, totaling at least $1.5 billion in upfront and equity payments and over $2 billion in additional milestone, royalty and other payments (Figure 1 & Table 3). It is worth noting, however, that for most Big Pharma companies their focus is on allogeneic approaches. Carolyn Green, Executive Director of Strategic Investments at Pfizer, has commented, “Autologous approaches do not fit the business model of Pfizer. CAR-T therapy will require very different manufacturing and distribution than traditional pharmaceuticals or antibodies – which are ‘one-to-many’ therapies. Pfizer has only one focus on CAR-T and that is the allogeneic approach, where an off-the-shelf product can be given as a ‘one-to-many’ strategy. We partnered with Cellectis on this.” In the past 2 years, three of the top 15 highest valued immuno-oncology partnerships were Big Pharma’s investments in CAR-T therapies [4]. Interestingly, two of these three partnerships are focused on allogeneic approaches and each is worth at least twice as much as the autologous partnership [4].
| Table 2 Initial public offerings of CAR-T companies 2011–2016. | |||
| Company | $ IPO (in millions) | IPO date | CAR-T approach |
| Bluebird bio | 101 | Jun 2013 | Autologous |
| Kite | 128 | Jun 2014 | Autologous |
| Bellicum | 140 | Dec 2014 | Autologous |
| Juno | 265 | Dec 2014 | Autologous |
| Cellectis | 228 | Mar 2015 | Allogeneic |
| Celyad | 100 | May 2015 | Allogeneic and autologous |
| Total | 962 | ||
| Only includes IPOs where the company had a CAR-T focus at the time of going public. IPO: Initial public offering. Source: Company press releases. | |||
Aside from the VC and Big Pharma investment deals discussed above, there have been over 75 additional partnership, acquisition and licensing deals done from 2012 through August 2016 worth at least another $650 million in disclosed upfront payments and $2 billion in additional milestone, royalty and other payments [Nelsen Biomedical Analysis]. The number of deals in this space has grown every year, from just three deals in 2012 to 35 in 2015. With already 26 deals as of September 1, 2016, the full-year 2016 numbers will likely reach at least the 2015 figure. These deals cover intellectual property, new approaches for creating CARs using gene-editing technologies, expansion to allogeneic approaches, combination therapies and more (Supplementary Table 1, nelsenbiomedical.com/cartdeals).
| Table 3 Big Pharma–CAR-T companies strategic partnerships 2011–2016. | |||||
| Company/ Institution | Big Pharma | Deal type | Deal value | CAR-T approach | Date |
University of Pennsylvania | Novartis | Exclusive global research and licensing agreement | Not disclosed | Autologous | Aug 2012 |
| Bluebird Bio | Celgene | Exclusive multiyear research and collaboration agreement and license agreement | Not disclosed | Autologous | Mar 2013 |
| Cellectis | Servier | Collaboration | Servier will pay $10 million upfront and up to $140 million for each of the 6 product candidates potentially developed | Allogeneic | Feb 2014 |
| Cellectis | Pfizer | Collaboration | Pfizer will pay $80 million upfront plus up to $185 million per product and royalties | Allogeneic | Jun 2014 |
| Transposagen | Johnson and Johnson | Collaboration and license agreement | Unspecified upfront; Johnson and Johnson will pay up to $292 million per treatment in milestones | Allogeneic | Nov 2014 |
| Kite Pharma | Amgen | Collaboration | Amgen to pay $60 million upfront and up to $525 million per product in milestone payments, plus royalties on sales and IP licensing | Autologous | Jan 2015 |
Ziopharm/ Intrexon | Merck | Strategic collaboration and license agreement | Merck to pay $115 million upfront, fee will be split equally between Intrexon and partner Ziopharm Oncology along with a commitment of up to $826 million more in milestones for the first two programs | Autologous & allogeneic | Mar 2015 |
| Bluebird Bio | Celgene | Collaboration | Celgene to pay $25 million | Autologous | Jun 2015 |
| Juno | Celgene | Collaboration | Celgene to pay approximately $1 billion, composed of an approximately $150 million upfront payment and approximately $849.8 million to purchase 9,137,672 shares of Juno's common stock at $93.00 per share | Autologous | Jun 2015 |
| Cellectis | Servier | Exclusive global license and collaboration agreement | Servier to pay $38.2 million upon signature and eligible for over $300 million of milestone payments, R&D financing and royalties | Allogeneic | Nov 2015 |
| Precision Biosciences | Baxalta | Collaboration | Baxalta to pay Precision $105 million upfront, followed by up to $1.6 billion in option fees and payments milestones and royalties | Allogeneic | Feb 2016 |
Juno | Celgene | Exercised option | Celgene will pay Juno a fee of $50 million and the companies will now share global development expenses for products in the CD19 program | Allogeneic | Apr 2016 |
Only includes strategic partnerships where Big Pharma companies invest in and obtain rights to CAR-T therapeutic programs. Not included, for example, the Roche and Genentech partnership with Kite Pharma in March 2016 to combine Kite’s CAR-T technology with Roche or Genentech’s small molecules. See Supplementary Table 1 for other deals like this. Payments are upfront only, Source: Company press releases. | |||||
Overall, the T-cell immunotherapy market is projected to be worth $30 billion by 2030, with CAR-T therapies likely to garner the most attention in the near future [5]. To understand the true commercial outlook it is necessary to examine the scientific evolution of CAR-Ts, their clinical applications and the barriers to commercialization.
The evolution of CAR-Ts
Monoclonal antibody-based therapies achieve anti-tumor activity by influencing T cells within the patient’s body through cell-intrinsic signaling. This is one way to modify the adaptive immune system and target cancer cells. At the National Institutes of Health, USA, Steven Rosenberg took a different approach by harvesting, expanding and re-infusing tumor-infiltrating lymphocytes from melanoma patients. While this technique laid the groundwork for future T-cell-based therapies, it showed limited efficacy. In addition, when the affinity of the T-cell receptor (TCR) was increased, safety issues emerged [6,7]. This therapy, like all based on TCRs, was dependent on the presentation of its target antigen in the context of a major histocompatibility molecule (MHC), which increases the complexity of engineering the TCR. The development of the CAR-T cells addressed this shortcoming of TCR-engineered T cells. The CAR combines the antigen-recognition portion of the B-cell receptor, which functions independently of MHC, with the T-cell intracellular signaling domain (CD3ζ) [8]. This allows the T cell to recognize any surface molecule to which an antibody can be made and results in a highly effective therapeutic tool, even in disease refractory to chemotherapy.
First-, second- & third-generation CAR-Ts: focus on improving effectiveness
In the most basic CAR construct, the functional sections are the variable portion of an antibody that is specific for the intended target (scFv or antigen recognition), usually a tumor-associated antigen, and the CD3ζ [9]. When the scFv binds to its target antigen, the CD3ζ initiates the TCR signaling cascade, thus activating the cytolytic function of the CAR-T [10]. The first-generation CARs (Figure 2) provide only one signal of the three required for activation of T cells [11,12]. As a result, T cells receiving stimulation through a first-generation CAR without accompanying co-stimulation often become anergic. This negatively impacts the ability of the cell to function effectively and its ability to persist over time.
These limitations were addressed by incorporating the signaling domain of a co-stimulatory molecule into the CAR construct (Figure 2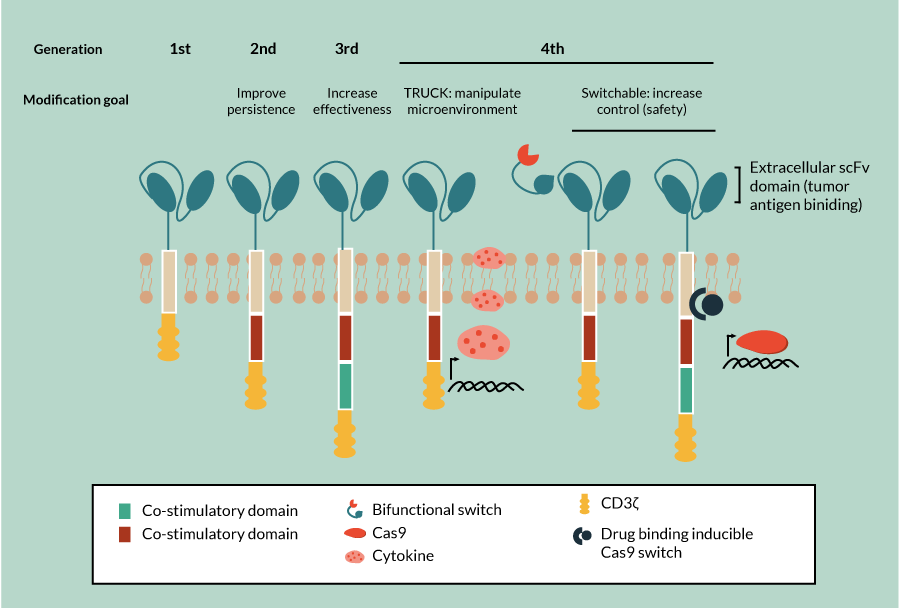
Next-generation CARs: to overcome suppression & improve safety
While second-generation CARs have achieved some significant preclinical and clinical successes, more improvements in safety and efficacy are needed. For example, even in the most successful CAR-T target to date, anti-CD19 CAR-T therapies, responses are variable. These therapeutics are more effective in treating CD19-expressing ALL than in other applications [15–18]. Reduced efficacy in some applications is due in part to suppressive effects that the tumor has on the CAR-T engineered cells. And these suppressive effects of the tumor microenvironment are more pronounced in solid tumors than in hematopoietic tumors. With the aim of correcting this suppressive effect, a new generation of CAR-T referred to as TRUCKs, deliver a ‘payload’ of immune-activating chemokines or cytokines (Figure 2). This enhances the effectiveness of the CAR-T through recruitment of additional immune cells to the tumor microenvironment and increased tumor killing via both these additional cells and the inflammatory cytokines themselves [19,20].
Regulatable CARs focus on improved safety
Many patients, approximately 55%, experience side effects related to cytokine release as a result of CAR-T therapy [21]. Cytokine release syndrome side effects are often mild, flu-like symptoms but can be severe, including hypotension, vascular leak, pulmonary edema and coagulopathy, leading to multi-organ failure and even death. Currently these complications are managed with corticosteroids and anti-IL-6 therapy [15]. Investigators are experimenting with incorporating elements into new CAR designs to allow them to be regulated, with the aim of increasing the safety of these therapies. Examples of recently developed safety measures include switchable CAR-T whose activity can be modulated by small molecules, and the inclusion of an inducible caspase 9 to allow directed apoptosis of the CAR-T (Figure 2) [22,23]. Additional approaches continue to be developed with the hope of improving safety through alternatives such as antibody-coupled T-cell receptor technology, in which the receptor on the T cell must be activated by a separately administered antibody and in this way titration of response can be achieved.
Clinical applications
The optimization of CAR-T constructs at the bench has translated not only to the success of these therapies in ALL, but also to the expansion of CAR-T clinical trials. From 2011 to 2015, trials using CAR-T therapies have grown in tandem with cellular immunotherapy, comprising 50% of all new cellular immunotherapy clinical trials and 25% of total cell therapy clinical trials overall (Figure 3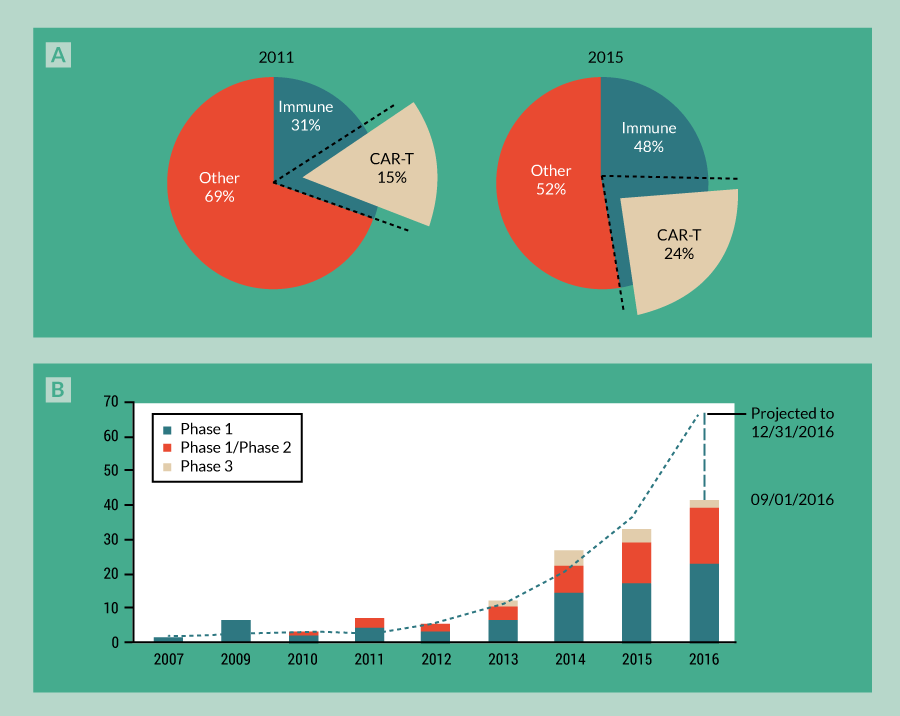
Blood cancers
CAR-T cells have shown extraordinary success in treating B-cell hematologic malignancies such as ALL, CLL and non-hodgkins lymphoma (NHL). Depending on the specific trial and indication targeted, overall response rates typically range from 40 to 94% [21]. Collectively, recent trials in patients with ALL, CLL or NHL show that on average 75% of patients experience a complete or partial response, with a majority (67%) showing a complete response [21].
Of these three blood cancers, the results in patients with refractory or relapsed ALL have been the most impressive. Most people have heard of the poster child of this game changer in the fight against this deadly disease, Emily Whitehead whose last ditch treatment by Dr Porter at the University of Pennsylvania has left the now 11-year-old cancer free for over 4 years. Since then, ALL patients treated with CAR-T therapies targeting CD19 exhibit complete response rates of approximately 90% [24], a remarkable improvement over other existing therapies for ALL, such as chemotherapy and allo-stem cell transplant (SCT) where response rates and durable remissions are <50% [25]. Notably, this 90% complete response success rate has been observed across all age groups, from pediatric to adult populations, and across independent trials taking place at different academic medical facilities [21,24,25]. The consistency in these successful outcomes in blood cancers is particularly profound, given that inconsistencies tend to arise due to the variety of protocols, infrastructure and clinical staff used across the clinical trials.
Other CAR-T therapies targeting CD19 to treat B-cell hematologic malignancies are equally impressive compared to response rates with current therapeutics. However, in comparison with ALL, CLL and NHL have yielded more variable response rates – between 40 and 85% overall response and 20 and 60% complete response [21,25]. While ongoing research is focused on understanding how to achieve more effective and consistent results using CAR-Ts for CLL and NHL, nevertheless this therapeutic approach for these malignancies appears to be more promising than those that are currently available.
Expansion to solid tumors
Driven by the success of the CD19-CAR in treating ALL, CLL and NHL, CARs specific for other cell-surface markers are being explored for the treatment of many different types of malignancies. While some target hematologic malignancies, such as the anti-CD38 CAR [26–28], others are focused on solid tumors. Examples include T cells expressing CARs specific for IL-11Rα and variants of CD44, which are targeted for head and neck, pancreatic, gastric, breast, cervical and colon cancers [29–31], or a CSPG4–CAR, which targets melanoma, triple-negative breast cancer, glioblastoma multiforme, mesothelioma, head and neck cancers, and osteosarcomas [32–34]. Clinical trials are ongoing for the treatment of neuroblastomas and osteosarcomas with a GD2-CAR-T therapy [35]. For neuroblastoma, early results look promising for the only program beyond blood cancers that has progressed to Phase 2 clinical trials (NCT02765243) [35]. Additional antigens are being tested preclinically for targeting solid tumors. Brain cancers, CNS cancers, gliomas, glioblastoma multiforme, and head and neck cancers are being targeted with an ErbB2-CAR-T cell [36]. Breast cancer responsiveness to CAR-T therapy is being explored using c-MET and carcinoembryonic antigen (CEA) CAR-T cells [37], while prostate cancers are being targeted with a prostate-specific membrane antigen CAR-T construct [38].
What’s the real potential for CAR-T in cancers beyond blood? One investor noted that: “We know that they work for B-cell lymphomas really well, but there’s going to need to be a suite of technical alterations for it to work in various other applications.” (Ben Auspitz, F-Prime Capital) Some barriers to success in solid tumors that need to be addressed through more research and technical improvements include:
- Identification of safe and effective antigenic targets for each tumor type
- Efficient localization of CAR-T cells within the tumor
- Neutralization of the immunosuppressive tumor microenvironment
Research to address these barriers is ongoing and varies by tumor type, currently ranging from early pre-clinical studies to clinical trials. As we discussed earlier, many antigenic targets are under investigation to expand CAR-T applications to other blood cancers and solid tumors. But most of these antigens are expressed on a variety of normal cell types. In solid tumors, targeting CARs to overexpressed antigens can lead to the destruction of healthy solid organ tissue, such as in the case of the off target-mediated mortality seen in HER2 CAR-T therapy for colon cancer [39]. So, clearly, safety is a concern and solid tumor-associated antigens must be carefully selected. A variety of approaches for optimizing CAR-T therapy for solid tumors range from directly administering CAR-T cells to the tumor, to combining CAR-T with small molecules that regulate cytokine and surface receptor responses, to transient CAR expression. These strategies are thoroughly summarized in two recent reviews in Molecular Therapy – Oncolytics [40] and the Journal of Cytotherapy [41].
CAR-T therapies beyond cancer
Further on the horizon, researchers have begun work to adapt CAR technology for the treatment of autoimmune diseases. For these applications the CAR is engineered to be reactive against self-antigens, but instead of being attached to a conventional T cell, it is attached to a regulatory T cell (Treg). In this way, it is hoped that tolerance against specific self-antigens can be achieved. This method has achieved some preclinical successes in mouse models of colitis and multiple sclerosis. CAR-Tregs for the treatment of colitis have been generated against multiple targets (2,4,6-trinitrophenol and CEA); both have shown specificity, improvement in survival and amelioration of inflammation-associated symptoms [42,43]. In the treatment of experimental autoimmune encephalomyelitis, which is a mouse model for multiple sclerosis, the CAR used was directed against myelin oligodendrocyte glycoprotein and produced a durable reduction in symptoms, even in the face of a second induction of disease [44].
Clearly, CAR-T technology holds promise in a variety of therapeutic areas when and where the highly specific nature of its antigen targeting can be leveraged. However, investors we spoke with said that only when additional antigens can be specifically targeted and show efficacy similar to that achieved with CD19 and B-cell lymphoma, will the field beyond blood cancers take off.
Challenges for CAR-T
There remain technical, clinical and commercial challenges that must be overcome for the widespread adoption and production of CAR-T therapy.
Technical challenges
Technically, some areas requiring additional work include:
- Identification of safe and effective antigenic targets for a variety of tumor types
- Optimizing CAR design for maximum efficacy
- Inclusion of appropriate safety controls into CAR design
These topics have already been discussed above. Briefly, to the first point, identifying tumor-specific antigens (not expressed by normal cells) will be key to avoiding on-target, off-tumor effects. However, even with effective antigens to direct CARs against, the tumor can down-regulate the antigen (antigenic escape), which can be detrimental to therapeutic efficacy. The prevailing approach to combatting antigenic escape is to target multiple antigens on the tumor, such as using CD22-CAR-T cells to treat patients who have relapsed with CD19-negative ALL following successful CD19-CAR-T therapy (clinical trials NCT02650414 and NCT02315612).
Often overlooked, but equally important to efficacy is how the cell therapy product is manufactured. Recent work has shown that “the method used for expanding T cells prior to infusion is an essential determinant of their in vivo efficacy” [45]. Companies need to give serious considerations to the way in which CAR-T cells are generated, as this can have significant impacts on efficacy. With manufacturing, enrichment of the ‘active ingredient’ may be achieved through a combination of:
- Improved protocols for transduction efficiency and cell selection
- Optimizing in vitro growth conditions to enhance outgrowth of the desired cell population
- Enrichment of the ‘active ingredient’
The effectiveness of CAR-T therapy may reasonably be expected to depend on the percentage of cells expressing the CAR of interest. In fact, comparing three different protocols for manufacturing CD19-CAR-T cells for clinical trials in ALL demonstrates the significant impact that the transduction efficiency can make in the results. Cells produced with one manufacturing protocol (see the first column in Table 4) [45] did not yield any responses during a Phase 1 clinical trial. Modifying both the vector and the transduction and expansion steps in a different manufacturing protocol improved efficacy significantly, giving a clinical response rate of 74% (see the second column in Table 4) [46]. In a third Phase I clinical trial for B-NHL, CAR-T cells produced from a central memory cell population using a more complex protocol achieved an 88% response rate. This was an improvement on a similar simpler protocol that utilized a bulk T-cell starting population and resulted in only a 74% response rate for B-ALL, which traditionally is the most responsive to CD19 CAR-T therapy (see column 3 in Table 4) [47,48]. Through these and other examples, the importance of optimizing transduction efficiency during manufacturing is clear. Accordingly, clinical protocols that do not include enrichment for cells expressing the target CAR may require higher initial transduction efficiencies. In addition, shifting manufacturing processes to optimize the input population of T cells with memory subsets, which have shown increased effectiveness [49], may further improve success rates.
| Table 4 Comparison of selected CAR-T manufacturing protocols. | ||||
| Protocol 1 | Protocol 2 | Protocol 3 | ||
Step 1 | Cancer Type | CD19+ ALL and CLL | CD19+ ALL | CD19+ NHL |
| Patient Population | Adult | Pediatric and Young Adult | Adult | |
| Cell Enrichment | All T cells | All T cells | Memory T Cells | |
| Step 2 | Transduction | Spinnoculation with Retronectin | Retronectin (No Spinnoculation) | Spinnoculation with Retronectin |
| Step 3 | Growth Factors | IL-2 | IL-2 | IL-2, IL-15 |
| Step 4 | Initial Expansion Step | Yes | Yes | Yes |
| Step 5 | Stimulation Bead Removal | Yes | Yes | Yes |
| Step 6 | Second Expansion Step | No | Yes | Yes |
| Vector | SFG (Retrovirus) | MSGV (Retrovirus) | HIV (Lentivirus) | |
% CAR Expression/ Product | 14–40% | 50–80% | 60–90% | |
Total Cell Fold Expansion | 100–700x | 10x | 600x | |
| Total Time Required | 2–3 weeks | 1–2 weeks | 3–6 weeks | |
| Clinical Results | NR | 74% RR | 88% RR | |
Optimizing growth conditions in vitro to improve results in vivo
A second area of focus should be the specific media, cytokines and atmospheric conditions provided during manufacturing. Small perturbations in these conditions can strongly influence the outgrowth of subpopulations within a heterogeneous input population. Optimizing the culture conditions to produce the required population through pre-culture enrichment protocols and control of media and atmospheric conditions will be an advantage in the production of adoptive cell therapies. Consider, for example, differing effects that co-stimulatory molecules may have on the metabolic needs of CAR-T cells. If the co-stimulatory molecule is CD28, the cell is pushed towards glycolysis and will require higher glucose levels. If the co-stimulatory molecule is 4-1BB, the cell is pushed towards oxidative phosphorylation and will require higher oxygen levels [50]. These are important considerations for both therapeutic design and manufacturing. Oxidative phosphorylation is more energetically efficient, so, for a hematopoietic target, 4-1BB may be the obvious choice for co-stimulatory molecule. However, for solid tumors existing in a hypoxic microenvironment, relying more on glycolysis through CD28 could be an advantage as it reduces the requirement for oxygen. When manufacturing these cells, yields of therapeutically effective CAR-Ts may be improved through careful monitoring and modification of oxygen and glucose levels throughout manufacturing.
Clinical challenges
Clinical challenges also pose a barrier to the successful commercialization of CAR-T. Three areas of particular concern outlined by investors we interviewed include:
- Defining and predicting potency of the active ingredient
- Standardization of practices across multiple clinical sites
- Developing clinical infrastructure
First, defining minimum potency standards is needed. Currently, a dose is defined as a number of cells administered. But a cell therapy dose consists of a heterogeneous mix of cells. As we discussed, selection of T-cell populations and efficient CAR transduction to these cells can significantly affect the potency of the cell product. However, as we are still defining important characterizations of these cellular therapeutics, qualitative standards defining requirements to obtain minimum clinical efficacy have not yet been established. Until then, demonstrating clinical equivalency when each batch is patient specific (n = 1) remains a significant challenge to success in clinical trials, regulatory approval and production on a commercial scale.
Second, clinical practices must also be standardized, such as the establishment of preparative regimens and treatments for adverse events. The importance of preparative regimens has recently been underscored by the deaths in Juno’s clinical trial for ALL using JCAR015. A balance must be struck in providing the correct lympho-depleted environment without causing adverse effects.
Lastly, the clinical infrastructure is needed to scale from single sites, where the majority of clinical trials are currently conducted, to the multiple sites necessary for successful commercialization of these therapies. Clinical trials now are typically performed at a single center with deep medical expertise. We need the clinical infrastructure, expertise and standardization to scale from one clinical site to many. Otherwise these therapies will not be successful in reaching larger patient populations and markets.
Commercial challenges
To gain perspective on the biggest barriers to the expansion of commercial opportunities for CAR-T therapies, we interviewed VC firms who actively invest in this space. While multiple areas for improvement were discussed, three key areas of focus emerged including:
- Availability of Good Manufacturing Practice (GMP) facilities
- Reduction in the cost of goods sold
- Application to bigger market opportunities
“Producing autologous CAR-T cells requires at least two unique types of facilities/processes. First, a virus manufacturer to produce retro- or lentivirus to modify the cells, and then also a cell manufacturing facility to process the patients’ cells, transduce them with the virus, and then expand, characterize and formulate them. Currently both types of facilities have long wait lists and additional ramp up is needed to scale supply.” (Michael Gladstone, Atlas Ventures). This 18-month waitlist exits both at commercial manufacturing sites as well as academic and institutional manufacturing facilities [Nelsen Biomedical Interviews]. This is an obstacle for those working to commercialize CAR-T therapy, and for investigators working on new approaches.
Cost of goods is an additional barrier that has been widely discussed [51–55]. The first FDA-approved dendritic cell (DC) therapy, Provenge®, was developed by Dendreon. DCs stimulate antigen-specific cytolytic and helper T-cell responses, which lead to the formation of immunological memory. This capability has been used as a strategy to develop autologous cancer vaccines. Despite the efficacy of Provenge, the commercial challenges posed by manufacturing resulted in Dendreon filing for bankruptcy. How then, can companies attempting to commercialize autologous CAR-T therapies avoid the same fate?
Fortunately, the industry has been working on developing more efficient and cost-effective solutions for manufacturing therapies where small-scale is full-scale. Shifts to disposable, small-footprint solutions as well as semi- or fully-automated, entirely enclosed manufacturing systems can reduce costs due to personnel, clean rooms and consumables. Notably, some of these automated, enclosed ‘plug-and-play’ systems in development may ultimately be able to manufacture cells from up to 30 donors in parallel. Putting this into perspective, one CEO of a cell therapy Contract Manufacturing Organization (CMO) noted that “many of our clients’ clinical programs are such that if they’re successful they will need to increase their current use from one to ten clean rooms a month, to 100 clean rooms a month. That’s simply not feasible. The concept of having a fully enclosed system that could process in parallel 30 patients at a time is very appealing.” (M Bamforth, Brammer Bio). At least ten closed manufacturing options are in development or on the market, with more improvements on the way [56]. The need to incorporate effective manufacturing has clearly been recognized by companies in the field, with increasing numbers of partnerships being executed to focus on manufacturing (Figure 4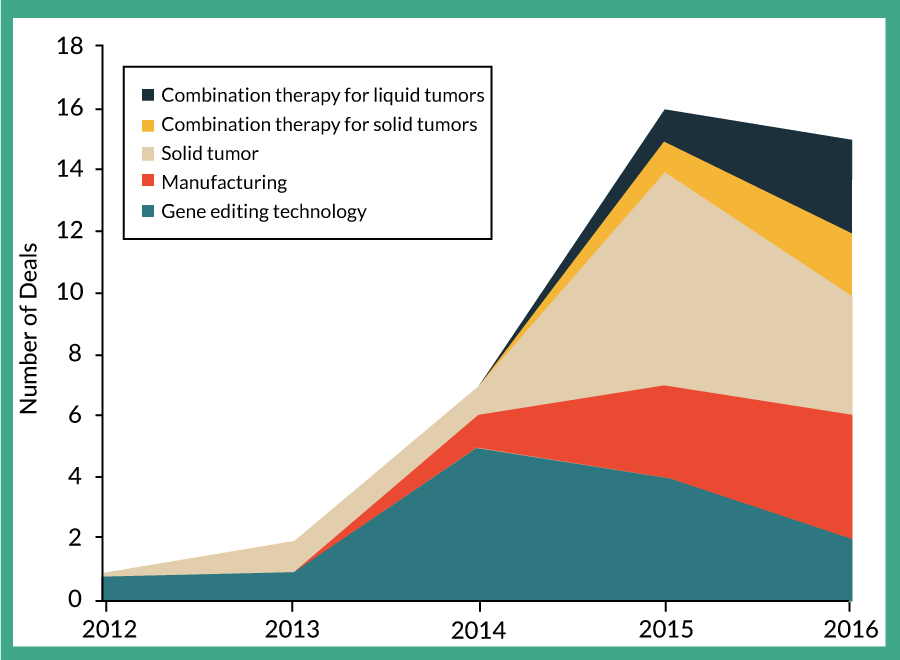
Of course, to avoid this issue of scale out, on solution is to develop allogeneic approaches, which are favored by Big Pharma (Table 3). There were encouraging clinical results from the first patient treated with Cellectis ‘off-the-shelf’ CAR-Ts [58–60]. It’s also worth noting that recently both Juno and Kite have made investments in developing off-the-shelf allogenic approaches (Supplementary Table 1, nelsenbiomedical.com/cartdeals).
Application to bigger market opportunities is the third point cited by investors as a significant challenge for CAR-T therapies. According to an experienced investor in this field, “most believe growth in the industry will stem from successes in solid tumors and that if we can’t figure solid out the CAR-T industry will stall”. While we are likely to see registration of CAR-T products for blood cancers such as ALL and NHL in the next few years, CAR-T for solid tumors is a long way behind. A variety of approaches are underway in an attempt to translate the successes of CAR-T therapies in treating hematologic malignancies to solid tumors, as discussed above. But it is worth noting again that the industry and clinicians will always adopt the simplest, safest solution that is effective. For solid tumors there are many approved therapeutics and more in development, from small molecules to monoclonal antibodies to combination therapies that provide some level of efficacy. A complicated, expensive cell therapeutic will only be adopted if it provides a significant improvement in clinical outcome compared to other options.
A quick look at recent deal types highlights the main challenges the industry is trying to address, from manufacturing, to easier CAR creation using gene editing, to improving efficacy with combination therapies (Figure 4). A more comprehensive view of the details of these recent deals can be found in Supplementary Table 1 nelsenbiomedical.com/cartdeals.
Moving the needle for cell therapy
The significant, startling successes of CAR-T therapies in patient populations with no therapeutic solution have provided the evidence needed for the cell therapy industry to gain interest from investors, the public markets and Big Pharma. However, the recent deaths in the Juno trial for ALL using JCAR015, and the exit of Novartis from the cell therapy industry, force us to revisit the practical and clinical realities of these types of therapies for broader market applications and adoption. Right now, everything about them is unwieldy, from collecting the initial donor material, to manufacturing individual batches of autologous cellular therapeutics, to developing pre-conditioning regimens.
Where are we now? The excitement in CAR-Ts has perhaps hit its peak until the additional technology, clinical and commercial infrastructure required matures (Figure 5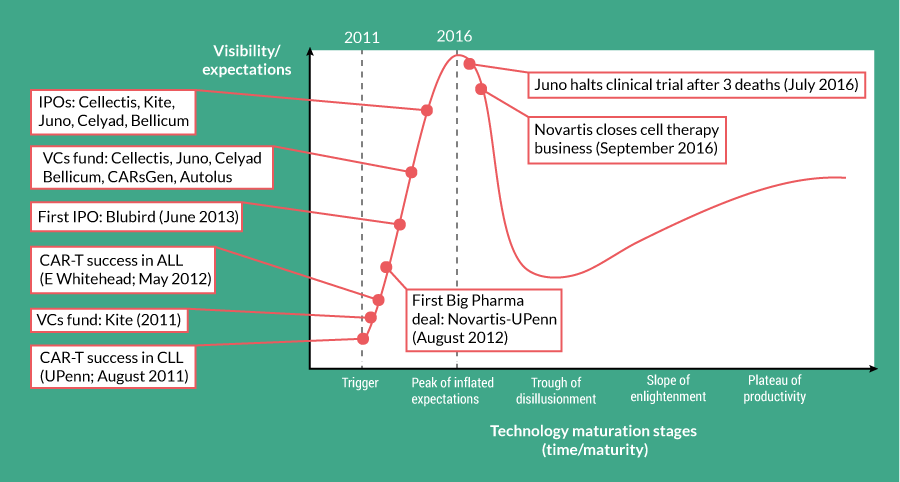
Fortunately, the past few years have seen increased activity and development of the supporting services and infrastructure needed for the entire cell therapy industry to move forward. Manufacturing solutions and services designed to reduce the cost of goods, clinical infrastructure and development of standard operating procedures to improve safety, efficacy and availability of treatment are just a few of the areas where investment and improvements are occurring. Simpler, cheaper methods for genetic modification for a variety of cells will also help to move the field forward. Comments Michael Gladstone of Atlas Ventures “If the [clinical and manufacturing] infrastructure can be built, it will lay the groundwork now for a better and more potent generation of therapies.” Beyond just advancing CAR-T therapeutics, “CAR-T therapy is also paving the way for development of other potentially effective cell therapy, including TILs, NK cells and γδ T cells.” (Shelly Chu, Abingworth). The advancement of even a single CAR-T therapy for a small patient population all the way to FDA approval will set a precedent that clears the way for the proliferation, adoption and commercialization of additional cell therapies.
Financial & competing interests disclosure
The authors have no relevant financial involvement with an organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock options or ownership, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Acknowledgements
The authors would like to thank Ben Auspitz of F-prime, Michael Gladstone of Atlas Ventures, Shelly Chu of Abingworth and Carolyn Green of Pfizer for sharing their time and insights.
This article was written with hope and anticipation for this future of curative solutions, and in memory of all those who have struggled waiting for this new age of cancer medicine, particularly William and Gloria Nelsen.
References
1. Brower V. The CAR T-Cell Race. The Scientist. 2015, April 1–7. Website
2. Venture capital funding for CAR-T. CB Insights 2016; 1–1.
3. Auer H. University of Pennsylvania and Novartis Form Alliance to Expand Use of Personalized T Cell Therapy for Cancer Patients [Internet]. 2016 [cited 2016 Jul 4]; 1–2. Website
4. Phillippidis A. Top 15 ImmunoOncology Collaborations. Genetic Engineering and Biotechnology News. 2016; 1–7. Website
5. Roots Analysis Business Research and Consulting. T-Cell Immunotherapy Market, 2015–2030. 2015 Oct; 1–4. Website
6. Rosenberg SA, Packard BS. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. N. Engl. J. Med. 1988; 319(25), 1676–80. CrossRef
7. Johnson LA, Morgan RA, Dudley ME et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009;114(3), 535–46. CrossRef
8. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl Acad. Sci. USA 1989; 86(24), 10024–8. CrossRef
9. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl Acad. Sci. USA 1993; 90(2), 720–4. CrossRef
10. Milone MC, Fish JD, Carpenito C et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009; 17(8), 1453–64. CrossRef
11. Kanagawa O. Three different signals are required for the induction of cytolytic T lymphocytes from resting precursors. J. Immunol. 1983; 131(2), 606–10. Website
12. June CH, Ledbetter JA, Lindsten T, Thompson CB. Evidence for the involvement of three distinct signals in the induction of IL-2 gene expression in human T lymphocytes. J. Immunol. 1989;143(1), 153–61. Website
13. Savoldo B, Ramos CA, Liu E et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Invest. 2011; 121(5), 1822–6. CrossRef
14. Abate-Daga D, Lagisetty KH, Tran E et al. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum. Gene Ther. 2014; 25(12), 1003–12. CrossRef
15. Davila ML, Riviere I, Wang X et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014; 6(224), 224ra25–5. CrossRef
16. Lee DW, Kochenderfer JN, Stetler-Stevenson M et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015; 385(9967), 517–28. CrossRef
17. Ramos CA, Savoldo B, Dotti G. CD19-CAR Trials. Cancer J. 2014; 20(2), 112–8. CrossRef
18. Ramsay AG, Clear AJ, Fatah R, Gribben JG. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: establishing a reversible immune evasion mechanism in human cancer. Blood 2012; 120(7), 1412–21. CrossRef
19. Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011; 71(17), 5697–706. CrossRef
20. Zhang L, Kerkar SP, Yu Z et al. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol. Ther. 2009; 19(4), 751–9. CrossRef
21. Park JH, Geyer MB, Brentjens RJ. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood 2016; 127(26), 3312–20. CrossRef
22. Ma JSY, Kim JY, Kazane SA et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc. Natl Acad. Sci. USA 2016; 113(4), E450–8. CrossRef
23. Budde LE, Berger C, Lin Y et al. Combining a CD20 Chimeric Antigen Receptor and an Inducible Caspase 9 Suicide Switch to Improve the Efficacy and Safety of T Cell Adoptive Immunotherapy for Lymphoma. PLoS ONE 2013; 8(12), e82742–10. CrossRef
24. Maus MV, June CH. Making Better Chimeric Antigen Receptors for Adoptive T-cell Therapy. Clin. Cancer Res. 2016; 22(8), 1875–84. CrossRef
25. Oluwole OO, Davila ML. At The Bedside: Clinical review of chimeric antigen receptor (CAR) T cell therapy for B cell malignancies. J. Leukoc. Biol. 2016,1–8. CrossRef
26. Mihara K, Bhattacharyya J, Kitanaka A et al. T-cell immunotherapy with a chimeric receptor against CD38 is effective in eliminating myeloma cells. Leukemia 2012; 26(2), 365–7. CrossRef
27. Mihara K, Yanagihara K, Takigahira M et al. Activated T-cell-mediated immunotherapy with a chimeric receptor against CD38 in B-cell non-Hodgkin lymphoma. J. Immunother. 2009; 32(7), 737–43. CrossRef
28. Bhattacharyya J, Mihara K, Kitanaka A et al. T-cell immunotherapy with a chimeric receptor against CD38 is effective in eradicating chemotherapy-resistant B-cell lymphoma cells overexpressing survivin induced by BMI-1. Blood Cancer J. 2012; 2(6), e75–3. CrossRef
29. Casucci M, Nicolis di Robilant B, Falcone L et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood 2013; 122(20), 3461–72. CrossRef
30. Dall P, Herrmann I, Durst B et al. In vivo cervical cancer growth inhibition by genetically engineered cytotoxic T cells. Cancer Immunol. Immunother. 2004; 54(1), 51–60. CrossRef
31. Huang G, Yu L, Cooper LJN, Hollomon M, Huls H, Kleinerman ES. Genetically Modified T cells Targeting Interleukin-11 Receptor -Chain Kill Human Osteosarcoma Cells and Induce the Regression of Established Osteosarcoma Lung Metastases. Cancer Res. 2012; 72(1), 271–81. CrossRef
32. Burns WR, Zhao Y, Frankel TL et al. A High Molecular Weight Melanoma-Associated Antigen-Specific Chimeric Antigen Receptor Redirects Lymphocytes to Target Human Melanomas. Cancer Res. 2010; 70(8), 3027–33. CrossRef
33. Schmidt P, Kopecky C, Hombach A, Zigrino P, Mauch C, Abken H. Eradication of melanomas by targeted elimination of a minor subset of tumor cells. Proc. Natl Acad. Sci. USA 2011; 108(6), 2474–9. CrossRef
34. Beard RE, Zheng Z, Lagisetty KH et al. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J. Immunother. Cancer 2014; 2(1), 1–11. CrossRef
35. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E. Antitumor activity and long-term fate of chimeric antigen receptor–positive T cells in patients with neuroblastoma. Blood 2011; 118(23), 6050–56. CrossRef
36. Pinthus JH, Fridman E, Dekel B et al. ErbB2 is a tumor associated antigen and a suitable therapeutic target in Wilms tumor. J. Urol. 2004; 172(4), 1644–8. CrossRef
37. Hombach A, Koch D, Sircar R et al. A chimeric receptor that selectively targets membrane-bound carcinoembryonic antigen (mCEA) in the presence of soluble CEA. Gene Ther. 1999; 6(2), 300–4. CrossRef
38. Ma Q, Gomes EM, Lo AS-Y, Junghans RP. Advanced generation anti-prostate specific membrane antigen designer T Cells for prostate cancer immunotherapy. Prostate 2014;74(3), 286–96. CrossRef
39. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol Ther. 2009; 18(4), 843–51. CrossRef
40. Newick K, Moon E, Albelda SM. Chimeric antigen receptor T-cell therapy for solid tumors. Mol. Ther. Oncolytics 2016; 3, 16006–7. CrossRef
41. Gad AZ, El-Naggar S, Ahmed N. Realism and pragmatism in developing an effective chimeric antigen receptor T-cell product for solid cancers. J. Cytother. 2016;1–11. CrossRef
42. Elinav E, Adam N, Waks T, Eshhar Z. Amelioration of colitis by genetically engineered murine regulatory T cells redirected by antigen-specific chimeric receptor. Gastroenterology 2009;136(5), 1721–31. CrossRef
43. Blat D, Zigmond E, Alteber Z, Waks T, Eshhar Z. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol. Ther. 2014; 22(5), 1018–28. CrossRef
44. Fransson M, Piras E, Burman J et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J. Neuroinflammation 2012; 9(1), 112. CrossRef
45. Hollyman D, Stefanski J, Przybylowski M et al. Manufacturing Validation of Biologically Functional T Cells Targeted to CD19 Antigen for Autologous Adoptive Cell Therapy. J. Immunother. 2009; 32(2), 169–80. CrossRef
46. MD DWL, MD JNK, MD MS-S et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a Phase 1 dose-escalation trial. Lancet 2015; 385(9967), 517–28. CrossRef
47. Wang X, Popplewell LL, Wagner JR et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood 2016; 127(24), 2980–90. CrossRef
48. Tumaini B, Lee DW, Lin T et al. Simplified process for the production of anti–CD19-CAR–engineered T cells. Cytotherapy 2013; 15(11), 1406–15. CrossRef
49. Wang X, Naranjo A, Brown CE et al. Phenotypic and Functional Attributes of Lentivirus-modified CD19-specific Human CD8+ Central Memory T Cells Manufactured at Clinical Scale. J. Immunother. 2012; 35(9), 689–701. CrossRef
50. June CH. 54th Annual J.S. and H.R. Blumenthal Memorial Lectureship: CAR T cells for treatment of cancer. 2016; 1–1.
51. Simaria AS, Hassan S, Varadaraju H et al. Allogeneic cell therapy bioprocess economics and optimization: single-use cell expansion technologies. Biotechnol. Bioeng. 2014; 111(1), 69–83. CrossRef
52. Hampson B. The Cost Of Success: Keeping Cost Of Goods In Check For Patient-Specific Cell Therapies. Life Science Leader. 2016 [cited 2016 Sep 19]. pp. 1–3. Website
53. Ratcliffe E, Thomas RJ, Williams DJ. Current understanding and challenges in bioprocessing of stem cell-based therapies for regenerative medicine. Br. Med. Bull. 2011; 100(1), 137–55. CrossRef
54. Preti B, Daus AM, Sumen C. Mapping Success for Commercial Cell Therapy Manufacturing. BioProcess International. 2015; 1–5. Website
55. Dodson BP, Levine AD. Challenges in the translation and commercialization of cell therapies. BMC Biotechnol. 2015; 1–15. CrossRef
56. Peterson B, Smith MJ, Nelsen B. Manufacturing Matters in Cell Therapy. Nelsen Biomedical 2016; 1–44. Website
57. Gray N. Cellectis’ T-cell therapy clears leukemia in second baby. BioPharma Dive 2016; 1–2. Website
58. Stanton D. Cellectis’ off-the-shelf CAR T-Cells treat 11-month old with leukaemia. BioPharma-Reporter 2015; 1–1. Website
59. Qasim W, Amrolia PJ, Samarasinghe S et al. First Clinical Application of Talen Engineered Universal CAR19 T Cells in B-ALL. Blood 2015; 126, 2046. Website
60. Brentjens RJ, Riviere I, Park JH et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011; 118(18), 4817–28. CrossRef
61. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2011; 365(8), 725–33. CrossRef
62. Grupp SA, Kalos M, Barrett D et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013; 368(16), 1509–18. CrossRef
63. Herper M. Juno Therapeutics Stops Trial of Cancer-Killing Cells After 3 Patient Deaths. Forbes 2016; 1–7. Website
64. Hallam K, Paton J. Novartis Dissolves Its Cell Therapy Unit, Cutting 120 Positions. Bloomberg 2016; 1–3. Website
Affiliations
Michelle J Smith*, Brittni M Peterson* & Barbara A Nelsen§
Nelsen Biomedical, Minneapolis, MN, USA
§Author for correspondence
barbara@nelsenbiomedical.com
*Contributed equally to the work.

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.