Holoclar: first of its kind in more ways than one
Cell Gene Therapy Insights 2016; 2(2), 183-197.
10.18609/cgti.2016.023
This article describes the regulatory pathway that enabled the translation of academic research and clinical experience of an ex-vivo expanded autologous stem cell-based treatment for limbal stem cell deficiency (LSCD) into a pharmaceutical product compliant with the European Union regulations of Advanced Therapy Medicinal Products (ATMP). Holoclar® was originally developed in Italy as a surgical procedure and used in more than 200 patients. Following the establishment of the EU ATMP Regulation EC 1394/2007, Holoclar development required that manufacturing was as per current Good Manufacturing Practice requirements and collection of retrospective clinical data was ICH-E6 and E3 compliant. Holoclar is an ex-vivo expanded autologous human corneal epithelial cells containing stem cells and is classified as “tissue engineered product”. Based on the evidence of quality and control of the manufacturing process, safety, efficacy and on a positive benefit–risk balance in 104 patients (72.1%) of 148 patients treated, Holoclar received conditional Marketing Approval in the EU. Holoclar is the first medicine approved in the EU for this rare eye condition that can result in blindness.
Holoclar® is the first stem cell product to be approved under medicines regulation in the Western world. It is also the first medicinal product to be approved based entirely on retrospective data in recent times to our knowledge, and the first advanced therapy medicinal product (ATMP) for an ophthalmic indication. In addition, it is probably the first product approved in the European Union (EU) after the adoption of ICH-E3 and ICH-E6 [1,2] based on data derived entirely from academic clinical research in vivo. Holoclar is manufactured by Holostem Terapie Avanzate s.r.l., a private–public partnership of Chiesi Farmaceutici S.p.A.
Holoclar comprises ex-vivo expanded autologous human corneal epithelial cells containing stem cells used for treating moderate-to-severe Limbal Stem cell deficiency (LSCD) caused by physical or chemical ocular burns. Such damage can lead to extensive/total loss of limbus. Since repair and restoration of damaged cornea is normally dependent on the presence of limbal stem cells, absence of it leads to conjunctivalization of cornea leading to loss of vision and significant discomfort. In the absence of limbal stem cells, surgical procedures such as corneal transplantation (keratoplasty) have a low success rate and short duration in benefit because of subsequent conjunctivalisation of the graft. By recreating normal corneal epithelium (including a sufficient amount of autologous stem cells), Holoclar was shown to successfully reverse the damage to cornea and restore healthy tissue and further facilitate the success of subsequent keratoplasty when needed (i.e., in cases of deep penetration of the burn injury in the corneal stroma).
Holoclar is manufactured starting from a small biopsy of 1–2 mm2 of undamaged limbus, taken from the patient’s unaffected eye (Figure 1). The biopsy is shipped from the ophthalmic center to Holostem TA s.r.l., Terapie Avanzate. Cells isolated from this sample are enzymatically dissociated and seeded as a single primary culture onto a layer of irradiated mouse feeder cells, with growth factors, that promote limbal stem cell (LSC) growth. Following primary culture expansion in media containing antibiotics, all cells are recovered and cryopreserved as an “intermediate cell bank” (ICB). After thawing an ICB, cells are seeded onto a fibrin matrix (secondary culture stage) which also contains the same type of mouse feeder cells; however, the media is changed to one that is antibiotic-free but still contains growth factors. The fibrin matrix provides a solid support on which the cells can be administered into the eye. After culture expansion, support discs made of fibrin carrying the autologous cells are prepared and packaged. Holoclar is then shipped and administered to the patient by grafting into the injured eye thus restoring the LSC reservoir.
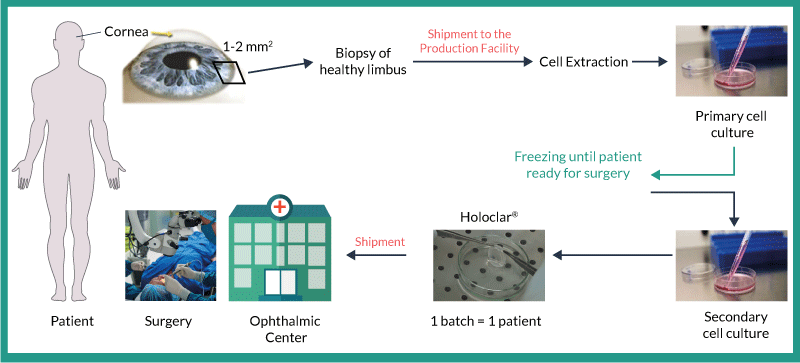
Figure 1. Manufacture and use of Holoclar.
Holoclar was approved entirely on the basis of retrospective data. This is virtually unheard of in modern times, as prospectively designed and conducted clinical trials are considered the gold standard for establishing convincing clinical benefit. However, in order to fully appreciate the specificity of this case, it is important to understand the background of the clinical problem and the environment in which the product evolved. LSCD due to physical/ chemical burns is a rare disease which is not expected to spontaneously heal with the restoration of the normal corneal epithelium and with limited treatment options. When an extensive fibrovascular pannus is present, the condition is irreversible and none of the available treatment options provides long-term benefit to the patient. The clinical studies were conducted at a time when a stem cell-based therapeutic agent was generally not considered as a medicinal product. Here we describe the story behind its origin and path to regulatory approval.
Disease Background
The normal human eye surface consists of cornea and conjunctiva separated by a specialized area called limbus. The basal layer of the limbus contains stem cells, which migrate centripetally to maintain corneal integrity and support corneal surface repair. The corneal epithelium consists of stratified squamous epithelium devoid of goblet cells and lacking any vascular support, as corneal cells extract nutrients from the lachrymal film. This key feature enables the corneal surface to be transparent (a property essential for vision) and immune privileged. Conversely, the surrounding conjunctival epithelium is highly vascular, non-transparent, and contains goblet cells that secrete mucin, contributing to the maintenance of normal tear film.
Alkali squirts is a common and devastating cause of the limbal stem cell deficiency, can affect workers in the construction and chemical industries but can also be a consequence of malicious assaults or other type of accidents. When such burns destroy the resident epithelial stem cell population in the periphery of the cornea, the physiological renewal of the corneal epithelium is compromised, as well as the repair process from the injury of the burn. In the absence of limbal stem cells, repair happens by proliferation of conjunctival epithelium invading the corneal surface, resulting in the generation of a superficial fibrovascular pannus and opacification, and leading to loss of vision (to complete blindness in the affected eye(s) in the most severe cases), inflammation and symptoms such as discomfort, pain, burning and photophobia.
Treatment options pre-Holoclar
Various surgical procedures have been used in the past to re-constitute the corneal surface. Simple excision of fibrous tissue and conventional keratoplasty are not sufficient to avoid recurrence of the fibrovascular pannus or tissue rejection, in cases characterized by limbal destruction. Until recently, the most frequently used surgical procedure for the treatment of this disease has been corneal transplant or lamellar (LKP) or penetrating keratoplasty (PKP). This in itself was found to have a low long-term success rate as it was subsequently determined that the transplant requires the presence of limbal stem cells for restoration of cornea. In the absence of limbal stem cells, keratoplasty is not sufficient to avoid re-epithelialization of the corneal surface by conjunctival cells.
Conjunctival limbal autograft (CLAU) provides epithelium restoration by using tissue from the contralateral healthy eye [3]. The limitation of CLAU is that this technique requires a large amount of tissue to be removed from the good eye and thus the potential donor risk [4]. Although few reports have shown consequences related to harvesting [5], patients are often unenthusiastic about having the “good” eye interfered with, together with the burden of responsibility felt by surgeons. Moreover, further harvesting of limbus following possible failure is not advisable. Finally, the CLAU is not applicable in case of bilateral lesions. In the allogeneic setting, a living relative (LR-CLAL) or a living unrelated donor (LNR-CLAL) or cadaver (C-CLAL) donates conjunctiva and limbal tissue. Kerato-limbal allograft (KLAL) utilizes cornea from a cadaveric donor to replenish the LSC niche [6]. However, donor epithelial cells do not survive in the long-term [7,8]. In the absence of demonstrated surviving donor cells, the explanation for clinical success is that patients with non-total limbal stem-cell deficiency have been treated, and the grafted allogeneic limbal cells might have induced modification of the microenvironment, and promoted proliferation of the patient’s own dormant stem cells, whose progeny gradually replaces donor cells.
Concept behind Holoclar
Keratinocyte stem cells govern the renewal of human stratified epithelia. These cells generate transient amplifying cells that terminally differentiate after a discrete number of cell divisions. The idea that stem cells had an important role in the successful repair of damaged epithelium was born out of experience with victims of third degree skin burns. Despite the best surgical technique, the results of skin grafting were highly variable. It was subsequently found that if there was an insufficient number of stem cells, the resulting skin tissue was not robust and easily damaged by trauma [9]. Following on from this discovery, it was investigated whether in the case of burn-induced corneal damage, the way to improve outcome would be through restoration of limbal stem cells. This was initially investigated by Kenyon and Tseng who carried out autologous limbal cell transplant by grafting large limbal fragments derived from uninjured eye [3]. However, this required rather large fragments, up to 30–50% of the total limbus, which may not always be achievable, might result in damage of the donor eye and is not suitable for repeated transplantations or bilateral lesions [4]. The possibility of growing limbal fragments in culture was soon recognized. The cultured cells were found to include a small population of stem cells detectable as holoclones (i.e., with 100% colony-forming properties and without differentiated cells). This led to the pioneering work by Pellegrini et al who carried out the first successful therapeutic application of limbal cultures for permanent regeneration of cornea in two subjects with unilateral ocular alkali burns [10]. This early success was followed by a larger study by Rama et al using culture of a biopsy of a small fragment (1–2 mm2) of limbus, cultivated under the same conditions as used for epidermal keratinocytes [11]. Confluent secondary culture, following trypsinization and embedding on a fibrin disc, were grafted over the cornea and limbus of the injured eye with considerable success in 14 of 18 patients.
Stem cells as medicines
Although the biological characteristics of stem cells are well established, their role as a medicinal product deserves further attention. Conventional medicinal products, be it small-molecule chemicals or more complex biologicals, tend to have direct and tangible mechanism of action, usually through a single mode such as pharmacological, metabolic or immunological effect. By contrast, stem cells have unparalleled versatility in the way they could potentially act as medicinal products, through various combinations of pharmacological, metabolic and immunological activities, regeneration, repair and replacement. However, using stem cells as a medicinal product is not without specific additional requirements. The manufacturing process is more complex and factors such as potency, characterization and consistency of production are some of the issues that need more attention, introducing new challenges compared to conventional medicines. In the specific case of tissue substitution, such as in LSCD, the mechanism of action of implanted cells comprises two main components:
- The implantation of mature or transient-amplifying cells in the treated site, which provide immediate substitution of the missing/damaged tissue; and
- The engraftment of stem cells (holoclones) that maintain spontaneous tissue regeneration and repair over time.
Therefore, the key factor driving durability of the treatment effect is the presence of a stem cell population in the product of sufficient amount and with a reasonable probability of engraftment in the correct niche.
Development of Holoclar as a medicinal product
Although regulation of medicines has been well established in Europe for decades, stem cells were not usually thought of (and regulated) as conventional medicinal products until quite recently, mainly falling within the legislative framework of tissue transplantations. Following the proof-of-concept study by Rama et al, the application of limbal stem cells for corneal damage was progressively extended into the practice of several eye clinics in Italy [11]. For this reason, it gained the national status of Consolidated Therapy treatment, as per “Gazzetta Ufficiale” 151/2007, in 2007 [12]. Subsequently, the ATMP regulation EC 1394/2007 was established and applied from 30 December 2008 [13]. This regulation provided, for the first time, the definition of a ‘Tissue engineered product’ and explicitly extends to ATMP the Directive 2001/83/EC which regulates medicinal products in the EU. The regulation implied that the “Consolidated Therapy” status would not be applicable after December 2012 (i.e., after a 5-year “grace period” to foster reconversion of existing products into ATMPs) and that a Marketing Authorization would be required to commercialize ATMPs from January 2013. The granting of a Marketing Authorization for an ATMP requires a scientific evaluation carried out by the European Medicines Agency on the type and amount of quality, preclinical and clinical data necessary to demonstrate the quality, safety and efficacy of the product required to commercialize ATMPs from January 2013. The granting of a Marketing Authorization for an ATMP requires a scientific evaluation carried out by the European Medicines Agency on the type and amount of quality, preclinical and clinical data necessary to demonstrate the quality, safety and efficacy of the product [14].
To provide the adequate level of quality manufacturing and Good Manufacturing Practice (GMP) compliance required by the pharmaceutical legislation, a spin-off company called Holostem TA was generated from the University of Modena and Reggio Emilia, Italy, where the laboratory of Prof. Pellegrini and Prof. De Luca (the inventors of the technique) were affiliated, with a traditional pharmaceutical company (Chiesi Farmaceutici, Parma, Italy) as the majority shareholder. Further development of Holoclar was taken over by Chiesi and Holostem starting from 2008. On reviewing the available data, the team at Chiesi came to the conclusion that there was sufficient existing data to demonstrate the value of Holoclar in the treatment of ocular burns. The reasons for this are as follows:
- A substantial number (219 in 21 centers) of patients suffering from this disease had already been treated by this time.
- The condition is sufficiently rare as to make this database robust in size, even if historical.
- Follow-up of between 2 and 10 years was available to support evidence of long-term benefit.
- End-points i.e., criteria for success were pre-defined and used in clinical practice to assess treatment outcomes.
- Same administration method i.e., surgical procedure was followed for all patients ensuring consistency in delivery.
- Similar post-surgical management including antibiotic and anti-inflammatory therapy as per keratoplasty protocols.
- Availability of photographic records of the affected eye for most patients provided objective evidence of the effect thus reducing the element of assessment bias, an inherent risk with retrospective data.
However, there were several hurdles to cross before the available data could be considered sufficient as pivotal evidence for a formal regulatory approval. These are as follows:
- Retrospective evidence is generally considered insufficient for regulatory approval. Indeed, to the knowledge of the authors, no medicinal product has been approved entirely on the basis of retrospective data in modern times.
- There was no control arm which would be typically expected in a confirmatory trial (only previous evaluation of the spontaneous evolution of pathology)
- It was not possible to demonstrate ICH-GCP compliance as this was a retrospective study that was not needed to be conducted as per clinical trial regulation pertaining to medicinal products.
- The manufacturing process needed to be made compliant to GMP which is a legal requirement as per regulations and converted to an industrial process to fit for commercial supply.
Regulatory strategy
Chiesi made an early decision to engage with regulators through European Medicines Evaluation Agency (EMEA; which was renamed as the European Medicines Agency [EMA] in December 2009).
The first task was to get Holoclar formally classified as a medicinal product. After taking advice from Innovation Task Force (EMEA/ITF), Chiesi applied to the Committee for Advanced Therapies (EMA/CAT) [15] and received a formal classification decision as a Tissue Engineered Product [16]. Chiesi also successfully applied for designation of Holoclar as an Orphan Medicinal Product by the Committee for Orphan Medicinal Products (EMA/COMP) [17]. The next task was to engage with the Scientific Advice Working Party (EMA/CHMP/SAWP) for formal consultation through a scientific advice (referred to as protocol assistance for orphan designated products) procedure [18] and agree on the general development plan, the key manufacturing quality aspects, and the clinical end-points to support the regulatory submission strategy.
The Pediatric Regulation envisages that an application for Marketing Authorization of a medicinal product is regarded as valid only if it includes results of all studies collected in compliance with an agreed pediatric investigation plan [19]. Accordingly, Chiesi applied for and obtained agreement on a Pediatric Investigation Plan (PIP) to defer pediatric studies, including waiver for children below 2 years, with the Pediatric Committee (EMA/PDCO) [20]. In the meantime, the company also acquired a GMP inspection certificate for the manufacture of Holoclar following the inspection of Holostem from AIFA, Italian regulatory agency, on behalf of EMA.
In addition, principles of ICH-E3 (study reporting requirements) [1] and ICH-E6 (Good Clinical Practices) [2] were to be applied to the retrospective data collection intended for regulatory review to the maximum possible extent, generating appropriate justifications and risk-assessment for any deviation.
Managing bias
One of the major disadvantages with a retrospective study is the possibility of introducing multiple systematic distortions (technically referred to as “biases”) in the assessment and interpretation of efficacy and safety outcomes. In fact, retrospective studies are known to be prone to biases in selection, reporting and investigator judgement, making evaluation of true benefit extremely difficult. For this reason, it is usually not possible to obtain regulatory approval based on retrospective data alone. However, in this specific case, data were re-evaluated in a blinded fashion by an independent assessor and the EU regulators acknowledged the potential value of existing clinical data in this rare condition and did not object in principle to the proposal to use retrospective data for marketing authorization via a Conditional Approval procedure, requiring compliance with post-authorization submission of supplementary prospective data, the latter as a routine requirement of conditional approval [21]. This approach required Chiesi to investigate the potential sources of biases in the data collection and to assess whether this had the potential to significantly influence the benefit-risk of the product. In this context, Chiesi proposed, and the SAWP agreed, on the generation of a prospective protocol (and statistical analysis plan) detailing how to collect and evaluate the retrospective data. The protocol was expected to cover all areas of a typical confirmatory study, including the concept, design, conduct and collection of data, as well as data management, analysis, and reporting system. Criteria for patient selection and for therapeutic success based on robust end-points were to be pre-defined (i.e., before actual data collection) and a statistical analysis plan put in place. The protocol was submitted and reviewed by the independent ethics committee of the participating centers (as an “observational study”) according to the AIFA determination on retrospective clinical studies [22]. Informed consent was to be obtained from all subjects whose data were to be collected.
Selection bias
In the case of patient selection, multiple concerns of potential biases should be considered. The most relevant risk is the way in which patients treated in consolidated clinical use were selected, which might not be representative of the future approved clinical indication. In fact, the product was researched and clinically tested outside of a traditional pharmaceutical development process and the access of patients to the treatment was based on strict eligibility criteria only in a group of cases. Furthermore, the homogeneity of selection was highly dependent on the practices of the clinical centers, with some centers applying a stricter protocol than others. On the other hand, the overall data could be considered more representative of the real-world use than that from a highly controlled clinical trial.
In order to address this potential bias and ensure consistency of data and applicability to future use, Chiesi decided to include all patients treated with autologous cultured limbal stem cell transplantation (ACLSCT ) and to stratify the treated patients in two distinct studies according to the level of stringency in selection of the patients by the treating physicians. Study HLSTM01 was prospectively designed to demonstrate the efficacy of ACLSCT in a cohort of 106 previously treated patients with defined characteristics. They were patients with LSCD resulting from ocular burns who had received the ACLSCT in two centers that consistently employed a standard protocol for surgical procedure, post-transplantation surveillance, and supportive pharmacological treatments. The two centers used the product in a context of clinical practice, but they were also pursuing the parallel aim of prospectively collecting research data, based on an “evolving protocol”. As compared to a GCP-compliant study, this approach was subject to ongoing changes reflective of limited initial procedural experience and subsequent accumulation of data. The selection criteria in study HLSTM01 were modelled on the original study and the ‘intention-to-treat’ principle was applied to data collection to account for any deviation from the protocol.
The second study (HLSTM02, with an additional 29 patients) sought to collect the safety data in all other patients who had received the ACLSCT in other centers. The patient population covered by the second study was considered more heterogeneous because it consisted of patients who had received the ACLSCT according to the same surgical procedure as in study HLSTM01, but with less control over eligibility (i.e., included patients with LSCD not caused by ocular burns) and with post-surgical clinical management and concomitant medication according to the established clinical practice in each center.
A second relevant source of potential bias relates to the probability of missing data due to the inability to include all patients previously treated. Here the concern is that the impossibility to include the overall population in the data collection process might lead to an undesirable consequence of selecting a sub-population of patients with unknown differences in clinical presentation or severity of the disease, exposure to risk factors, or outcomes of interest compared to the future to-be-treated population. Medical Surveillance Bias and Berkson Bias are typical examples of this type of bias [23] and the risk of incurring this bias is very high as information on treated patients is usually not exhaustively stored in a single (or limited number of) repository(ies). In the case of Holoclar, the highly individualized nature of the treatment allowed the sponsor to identify the total number of subjects treated to be 219 between 1998 and 2007 (i.e. up to the time of starting the collection). Of these, only 135 (61.6%) were available for the efficacy and safety analyses in support of the Marketing Authorization Application. Data for the remaining 82 patients were not available as the investigators declined the invitation to participate and release the clinical data. A more detailed investigation was undertaken to evaluate the risk that this could invalidate the available evidence. Two published studies [9,24] included 25 of these remaining 82 patients (12 and 13 respectively). The results published in these two studies were comparable to the available data of the HLSTM01 and HLSTM02 studies and supported the positive clinical benefit. It could be concluded that the missing data related to 82 patients did not negate the conclusions on clinical benefit based on the data available on the other 135 patients.
In response to questions raised by initial regulatory evaluation, Chiesi conducted several sensitivity analyses and sub-group analyses. The results suggested a very low probability that bias would have played a significant role in selection.
Information bias
The other important source of bias is distortions or mistakes in the collection of information. To address this, Chiesi followed the recommendations on the quality system that needs to be put in place in terms of data collection, traceability, and analysis as described in the ICH-E6 Guideline on Good Clinical Practices (GCP). Although GCP have been conceived for prospectively collected data, the two retrospective studies were conceived and designed following detailed discussions and considerations of all aspects in order to comply as much as technically feasible with such guidance in design and execution. In fact, the clinical study protocols and a data management system were defined a priori before starting data collection at the clinical sites, including a dedicated Case Report Form (CRF). In addition, a consistent approach to data collection and analysis/review was applied (including training of investigators) and a clinical contract research organization was appointed for data source verification. A Statistical Analysis Plan was prepared for each study before database lock and results have been collected in two Clinical Study Reports, structurally compliant with ICH-E3 guidelines.
However, the intrinsic quality of the existing data collected in hospital systems and clinical health records is usually insufficient in providing consistency in the way key variables were originally measured and collected, often making it impossible to adequately verify the data. In this sense, the uniqueness of the Holoclar case relies on a crucial element: the investigators of the two sites included in the HLSTM01 study had both participated in the early clinical testing of the product. During that phase, a data collection form was generated to prospectively gather the key outcome assessment variables, including corneal neovascularization, stability of the epithelium, symptoms, visual acuity etc. This data collection form became part of their outpatient health record and filled in at each patient’s visit as standard practice. The study variables were modeled around the available data. In addition, the two investigators collected pictures of the eyes of many treated patients at different occasions. This enabled the re-evaluation of the degree of corneal neovascularization (which was the key element in the definition of the primary efficacy end-point of the study) by an independent assessor in a blinded fashion. The external evaluation was reported as a secondary endpoint of the study and enabled to verify the absence of a significant degree of reporting bias.
Finally, an extensive analysis of missing data (including several sensitivity tests) revealed that the degree of this additional potential issue was minimal and had virtually no impact on the overall results.
Table 1 provides an overview of the identified potential sources of bias and of the steps taken to minimise them in the HLSTM studies. At the conclusion of this exercise, it was clear that this peculiar case of retrospective data collection could generate reliable, high quality data adequate to provide sufficient information on the efficacy and safety of the product so that a benefit–risk evaluation could be conducted. Nevertheless, some development gaps are remaining. The development and regulatory strategy of Holoclar was therefore built around the characteristics of the disease (rarity, severity), the strengths of the collected data (amount, quality) and the development gaps, committing to the collection of further data after approval.
| Table 1. Action to minimize bias in retrospective clinical study designs. | |
|---|---|
| POTENTIAL BIAS | CONTROL AND MINIMIZATION IN HLSTM STUDIES |
| Lack of representativeness | Inclusion of all available patients in two different studies |
| Sensitivity and sub-group analyses (consistency across sub-groups) | |
| Planning of a prospective study and a registry-based data collection | |
| Missing patients | Systematic analysis of the literature |
| Data quality | GCP-like approach to data collection (investigators training and data collection monitoring by an external CRO) |
| Missing information | Analysis of missing information at baseline and end-point visit |
| Open assessment | External re-analysis of eye photographs by an independent assessor |
Clinical Evidence
Dose selection
It is noteworthy that, unlike a conventional medicine, no formal dose-response study was possible because of the nature of the product and administration. As an autologous product for a single surgical administration, where healthy volunteer study is not possible, dose selection relied on measurement of the size of the patient’s cornea and set specifications of the drug product such as physiologic cell density and potency.
Efficacy end-points
The primary efficacy endpoint of the study was a composite endpoint based on evidence of neovascularization and epithelial defects at 12 months post-intervention. The transplantation was considered successful if it achieved the following:
- A superficial corneal neovascularisation classified as ‘None’ (no vessel penetration) or ‘Mild’ (vessel penetration 1 quadrant without central cornea involved); and
- Epithelial defects classified as ‘None’ (no fluorescence staining) or ‘Trace’ (minimal superficial staining, pooling with light and/or late staining).
The criterion for success of the treatment was defined as positive outcome in more than 50% of treatments as this was considered the minimal effect of clinical relevance in the management of patients.
Secondary endpoints included change from baseline to 12 months post intervention in symptoms (pain, burning and photophobia), inflammation and visual actuity; superficial corneal neovascularisation; number of LSCTs and number of successful keratoplasties after LSCT.
Good Clinical Practice
Since the clinical studies were not conducted as per the norms of GCP, this aspect needed resolution. Following visits to clinical sites, under the mandate of the EMA, the GCP inspectors of National Agencies (Italy and Germany) concluded there were no critical deviations that would have invalidated the validity of clinical data and thus could be used for assessment for the purpose of Marketing Authorization.
Regulatory conclusions on benefit–risk
The dataset provided in support of this application was considered unique and relatively large considering the rarity of the target condition. Despite the inherent disadvantages of a retrospective, uncontrolled and non-randomized study design, the quality of the dataset was considered adequate and the study results were compelling in demonstrating a clinically relevant benefit, considering that LSCD is a condition that would not spontaneously improve. Treatment with Holoclar allowed successful ocular surface reconstruction with improvements in symptoms and visual acuity in patients with moderate-to-severe LSCD. Clinically relevant outcomes were observed both in patients with and without deep stromal injury. These favourable effects were considered by the CAT to outweigh the risks of mainly ocular adverse reactions, which were generally manageable [25]. By providing training to treating physicians including a detailed treatment protocol recommending effective anti-inflammatory and anti-infective prophylaxis, it is expected that risks of adverse reactions can be further reduced.
In conclusion, the benefit–risk balance for Holoclar in the treatment of adult patients with moderate to severe limbal stem cell deficiency (defined by the presence of superficial corneal neovascularisation in at least two corneal quadrants, with central corneal involvement, and severely impaired visual acuity), unilateral or bilateral, due to physical or chemical ocular burns and with a minimum of 1–2 mm2 of undamaged limbus for biopsy, was considered favorable.
Conditional Approval
Chiesi applied for a Conditional Approval on the basis of Holoclar being an orphan product with demonstrable clinical benefit and the benefit to public health through immediate availability for this unmet medical need outweighed the limitations of evidence [21].
Furthermore, Chiesi was willing to fulfil obligations required under this procedure [21]. As a result, Conditional Approval was given and the following obligation was agreed as post-authorization measure:
Multinational, multicentre, prospective, open-label, uncontrolled interventional study (HLSTM03) to assess the efficacy and safety of autologous cultivated limbal stem cells grafting for restoration of corneal epithelium in patients with limbal stem cell deficiency due to ocular burns. The results of the study are to be submitted by December 2020 [25].
Additional risk minimization measures were agreed with the Agency as further conditions and requirements of the Marketing Authorization that are:
Long-term safety and efficacy follow-up after autologous cultivated limbal stem cells grafting for restoration of corneal epithelium in patients with limbal stem cell deficiency due to ocular burns (HLSTM03-FU)
Post-authorization Registry entitled ‘Long-term safety after Holoclar implant for restoration of corneal epithelium in patients with limbal stem cell deficiency due to ocular burns: observational study of routine clinical practice.’
A Conditional Approval is a procedure to expedite the availability of medicines in certain categories (i.e., the orphan drugs) to treat serious diseases and fill an unmet medical need. Provided a benefit–risk balance of the product have already been determined to be positive the medicine may be approved on the basis of surrogate markers and/or other less complete data than is normally the case but subject to specific obligations. However, the benefit– risk balance of the product should have already been determined to be positive. The Conditional Approval is a limited license, which can be converted to full license subject to applicants fulfilling their obligations and the review of additional evidence, failing which the authorization could be withdrawn. The conditional approval by the EMA is equivalent to the accelerated approval by the FDA [26], both approvals being based on a degree of uncertainty arising from use of less than the usually complete data and/or surrogate markers. The two major differences between the two agencies are: (a) the EMA requirement for re-assessment of the conditional approvals for their benefit–risk on an annual basis and (b) the EU regulation providing for financial penalties in case of non-compliance with commitments agreed during conditional approval.
Conclusions
Holoclar as an approved medicinal product in the EU is unique in many ways and can provide valuable lessons for future cases. First, it demonstrated that it is possible to receive a positive opinion and regulatory approval on the basis of complete retrospective data. However, this can be extremely challenging and likely to be an exception rather than a rule, as there are many potential hurdles to be overcome before success can be achieved. Although it appeared that sufficient data were available and the scientific basis to support a positive clinical benefit was strong, inadequacies in methodological matters could have potentially negated these gains and adversely affected the outcome. In addition, there were several other unusual features in Holoclar development. No dose-finding study was possible. The primary end-points were based on anatomical features (epithelial defects and superficial neovascularization) rather than clinical outcomes as these represent the most established and direct consequences of stem cell deficiency. Improvement in visual acuity was a secondary endpoint as, while being a key clinical outcome of the condition, is not dependent exclusively on LSCD. Indeed, severe visual impairment and blindness (associated with ocular burn injury) may result from damage of multiple levels of the eye, including the corneal stroma. In such cases, successful restoration of LSC function would not necessarily lead to an improvement in visual acuity, as the stroma would still be scarred. However, treatment with Holoclar would allow for subsequent keratoplasty, which would replace the scarred stroma and restore visual acuity. Therefore, an improvement in visual acuity was not used as a primary endpoint, as only a subset of patients (i.e., those without stromal damage) could be expected to demonstrate such an improvement.
There were substantial missing data as many centrers declined to share data. Despite these obstacles, by thorough planning, including several regulatory interactions and execution of a viable regulatory strategy it was possible to achieve a successful outcome. Maintenance of high quality records, which was a pre-requisite for retrospective studies, by the academic investigating centers made it possible. Foremost however, the key element leading to the approval was the evidence of a clinical benefit reflected by the restoration of a stable corneal epithelium with resolution of epithelial defects, regression of corneal vascularisation, and absence of conjunctivalisation. Long-term data up to 10 years, although limited, suggested persistence of the effect. Overall, this was a convincing outcome considering that LSCD would not be expected to improve spontaneously.
On their part, the regulators were willing to keep an open mind throughout the interactions and were supportive where possible. Despite the entire evidence having been derived from retrospective data, emphasis was given to the strength of scientific evidence rather than on the absence of traditional methodological approach and a formal prospective development plan. It is important to note that the implementation of manufacturing under GMP, the post-approval commitments (conduct of a post approval confirmatory study and study registry) and the commitment to train the relevant staff in the clinical centers that will use Holoclar were key elements driving the decision to grant the conditional MA [25].
The Holoclar experience exemplifies the value of positive interaction between academia, industry and regulators in a highly complex area such as stem cell therapy in a rare and unmet medical need leading to a success story.
Financial & competing interests disclosure
GM, DA and MT are employees of Chiesi Farmaceutici, Italy. GP and MDL are shareholders Holostem Terapie Avanzate and Consultants of J-TEC. Dr Goplan Narayan has supported the authors in the writing of the manuscript.
Acknowledgements
The authors wish to thank the clinical investigators and their teams for participating to the retrospective clinical studies HLSTM01 and HLSTM02: Gian Maria Cavallini, Alessandro Lambiase, Claudio Macaluso, Marco Nardi, Augusto Pocobelli, Leopoldo Spadea, Achille Tortori, Carlo Enrico Traverso and Paolo Vinciguerra.

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.
References
1. CPMP/ICH/137/95 Structure and Content of Clinical Study Reports, 1995. Website
2. CPMP/ICH/135/95 Guideline for Good Clinical Practice, 1996. Website
3. Kenyon KR, Tseng SC. Limbal allograft transplantation for ocular surface disorders. Ophthalmology 1989; 96(5): 709–22. CrossRef
4. Baylis O, Figueiredo F, Henein C, Lako M, Ahmad S. 13 years of cultured limbal epithelial cell therapy: a review of the outcomes. J. Cell Biochem. 2011;112(4): 993-1002. doi: 10.1002/jcb.23028. CrossRef
5. Jenkins C, Tuft S, Liu C, Buckley R. Limbal transplantation in the management of chronic contact-lens-associated epitheliopathy. Eye (Lond). 1993; 7(5): 629–33. CrossRef
6. Bakhtiari P, Djalilian A. Update on limbal stem cell transplantation. Middle East Afr. J. Ophthalmol. 2010; 17(1): 9–14. CrossRef
7. Henderson TR, Coster DJ, Williams KA. The long term outcome of limbal allografts: the search for surviving cells. Br. J. Ophthalmol. 2001; 85(5): 604–9. CrossRef
8. Daya SM, Watson A, Sharpe JR, Giledi O, Rowe A, Martin R, James SE. Outcomes and DNA analysis of ex vivo expanded stem cell allograft for ocular surface reconstruction. Ophthalmology 2005; 112(3): 470–7. CrossRef
9. De Luca M, Pellegrini G, Green H, Regeneration of squamous epithelia from stem cells of cultured grafts. Regen. Med. 2006; 1(1): 45–57. CrossRef
10. Pellegrini G, Traverso CE, Franzi AT et al. Long-term restoration of damaged corneal surfaces with autologous cultivated corneal epithelium. Lancet 1997; 349(9057): 990–3. CrossRef
11. Rama P, Bonini S, Lambiase A et al. Autologous fibrin-cultured limbal stem cells permanently restore the corneal surface of patients with total limbal stem cell deficiency. Transplantation 2001; 72(9): 1478–85. CrossRef
12. Gazzetta Ufficiale Repubblica Italiana 151, 02.07.2007, Annex 1.5 Website
13. Regulation (EC) No 1394/2007 of the European Parliament and of the Council on advanced therapy medicinal products and amending Directive 2001/83/EC and Regulation (EC) No 726/2004. OJ L324, 121–137 (2007).
14. Regulation (EC) No 726/2004 of the European Parliament and of the Council on laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency. OJ L 136, 1–70 (2004).
15. EMA/CAT Procedural advice on the provision of scientific recommendation on classification of advanced therapy medicinal products in accordance with article 17 of regulation (EC) no 1394/2007. Website
16. Summaries of scientific recommendations on classification of advanced-therapy medicinal products. Website
17. Holoclar Public summary of opinion on orphan designation (2009). Website
18. EMA Guidance for applicants seeking scientific advice and protocol assistance (2010). Website
19. Regulation (EC) No 1901/2006 of the European Parliament and of the Council of 12 December 2006 on medicinal products for paediatric use and amending Regulation (EEC) No 1768/92, Directive 2001/20/EC, Directive 2001/83/EC and Regulation (EC) No 726/2004. OJ L 378, 1–19 (2007).
20. Holoclar Paediatric investigation plan Public summary of opinion (2015). Website
21. Regulation (EC) No. 507/2006 of the European Parliament and of the Council of 29 March 2006 on the conditional marketing authorisation for medicinal products for human use falling within the scope of Regulation (EC) No 726/2004 of the European Parliament and of the Council. OJ L 92, 6–9 (2006).
22. Determinazione AIFA 20 Marzo 2008 Linee guida per la classificazione e conduzione degli studi osservazionali sui farmaci. Gazzetta Ufficiale Repubblica Italiana nr 76, 31 Marzo 2008.
23. Daniel Westreich. Berkson’s bias, selection bias, and missing data. Epidemiology 2012; 23(1): 159–64. CrossRef
24. Marchini G, Pedrotti E, Pedrotti M et al. Long-term effectiveness of autologous cultured limbal stem cell grafts in patients with limbal stem cell deficiency due to chemical burns. Clin. Exp. Ophthalmol. 2012; 40(3): 255–67. CrossRef
25. Holoclar, European Public Assessment Report (2015). Website
26. FDA Guidance for Industry Expedited Programs for Serious Conditions – Drugs and Biologics. Website
Affiliations
Chiesi Farmaceutici S.p.A., Italy