Centralized or decentralized manufacturing? Key business model considerations for cell therapies
Cell Gene Therapy Insights 2016; 2(1), 95-109.
10.18609.cgti.2016.012
The choice of manufacturing strategy for cell-based therapeutics is one that is best made early in product development. Expense and delay may result from any additional bridging studies following changes to manufacturing process design late in development. The chosen strategy will be strongly influenced in turn by the preferred business model. Business models may favor either centralized or distributed manufacture. There are advantages and disadvantages to each and variants may be suitable in certain circumstances. An appropriate choice depends upon a combination of regulatory, economic and supply-chain factors. In this article the factors are examined and described in the context of hypothetical examples. In general the degree of decentralization will depend on a balance of manufacturing features. The investment risk of building a centralized facility at the projected capacity, the cost of managing quality and the cost or quality implications of long-distance cold supply chains must be considered. No single business model will suit all cases. For any innovation the decision must be based on an operational analysis at the projected capacity required.
Submitted for review: Dec 1 2015 Published: Mar 21 2016
Cell-based therapies (CBTs) offer new ways to treat many medical conditions. In some cases the conditions could only otherwise have been managed using palliative care. CBTs are headline grabbing and the interest in their potential can obscure the remaining challenges to commercial-scale manufacture. CBTs have peculiar challenges that arise from the scale and patient-specificity of the product [1]. Their manufacture is managed under the regulations covering medicinal products; however, the complexity of CBTs has led to enhanced regulatory guidance and controls.
This article focuses on commercial-scale manufacture that requires compliance with Good Manufacturing Practice (GMP) [2,3]. (Products can be made in the EU on compassionate grounds under exceptions to GMP and many advances in treatment have so far been applied by making them at limited scale as ‘exemptions’ or ‘specials’ [4,5].)
The research and development of medicinal products is cash intensive with much research being carried out by large companies with sufficient resources to endure the intervals between successful drug candidates. Manufacture of medicinal products is usually based on the principle of scaling up in a centralized manufacturing facility. The facility will contain plant and equipment suitable for a well-established series of unit operations but that may be reconfigured in different ways to meet the needs of new products. The behavior of the unit operations is well understood by biochemical and chemical engineers. The plant and equipment is familiar in the form of large-scale stirred tank reactors, solvent-recovery columns and batteries of liquid-transfer piping. In such an environment the cost of maintaining and renewing the factory is spread over many product types; there is ‘economy of scope’ as well as ‘economy of scale’. New designs for the unit operations take many years to become established in practice.
By contrast CBTs are frequently introduced by small, often start-up, companies or by researchers who are not employees of the larger companies [6]. In the experience of the author it is unusual for these expert entrepreneurs to be experienced in manufacturing research. Their inventions require deep knowledge of the product characteristics rather than manufacturing operations. As a result the products may reach quite a mature stage in development before a decision is made to manufacture at large scale or to offer the innovation to an established company. When the commercialization decision is made, manufacture will often be based on research-scale equipment. A business that wishes to commercialize the CBT now needs to resolve a dilemma; and the dilemma arises from two factors. The first factor is the need to avoid unnecessary outlay before the CBT has proved that it can generate satisfactory revenue. The second factor is that the CBT may not be suitable for scale-up using the existing plant. The business may decide to resolve the dilemma by continuing to manufacture stock for clinical trial using the research-scale process by which the concept was proved at pre-clinical scale. However, for any medicinal product it is essential to understand the features of the manufacturing method (the Critical Process Parameters or CPPs) which exercise the most influence upon the important product properties (the ‘Critical Quality Attributes’ or CQAs). The CPPs must then be conserved during process development and scale up. For a small-molecule pharmaceutical the impact of any significant changes in the manufacturing method can be examined and justified relying mainly on prior knowledge of the chemistry and analysis [7]. For such products the conservation of the CQAs can be asserted with a high level of confidence. The scaling factors for the process are much better understood and there are fewer mechanisms by which the product can be affected without the resultant change appearing in the results of analysis. In regulatory terms the batches made at a later stage of process development are ‘comparable’ to the batches upon which proof of concept and efficacy was based [8]. For CBTs as for biotechnology products generally the impact of the process changes is much less well understood and analysis alone is unlikely to reveal evidence that is compelling proof of comparability [9–11]. Additional assurance of quality, safety and efficacy is required in the form of confidence based upon continued compliance with the established manufacturing process [9].
A hypothetical product is given as an example: a treatment based upon allogeneic cells to treat a non-life-threatening condition, such as inflammatory damage to joints. Let us suppose that the research work has been accomplished at small scale and three key commercial features have been proven:
1. The cells expand to commercially-useful numbers with retention of the phenotype and key quality attributes.
2. The cost of producing the required numbers of cells is satisfactory compared with the market price or reimbursement that will be achieved.
3. The market is of a size that justifies the investment required to launch the product.
These questions have been answered by expanding the cells at laboratory scale. At each passage a sub-set of the cells has been taken onto the next stage. The variable costs of production have been calculated; this was based largely on the dominant costs of the medium and, perhaps, the disposables. An allowance for labour was made along with estimates of operating costs for a suitable facility. Forty-layer cell factories have been named as the method of manufacture at commercial scale. The hidden factor in this popular treatment is ‘operability’ [12]. Some features of manufacture do not scale well–materials movement on a factory floor is such a feature. Manufacture using tens of thousands of cell factory units per annum will place unrealistic demands on cleanroom management and worker engagement. A change at this stage in development, for example to microcarrier culture, departs from the manufacturing method used during preclinical studies. To demonstrate that the product from the revised process retains its CQAs will require bridging studies with no guarantee of success.
If the product is of high value, and the market is low volume, it may be possible to proceed using the research-scale process. For all other products one of two routes must be taken. The first route is to repeat the early work using the process that is to be used at commercial scale. The second route is to begin the research with a clear vision of how and where the product will eventually be made and to adopt a process that will satisfy the commercial manufacture.
The second route is less expensive and brings the product to market more quickly. However, it requires an early choice. This choice can be made following careful examination of the logistics that are needed to supply the goods and the cost implications of these. The scope of the logistics should include manufacture, transit to the place of use and the arrangements for hand-off to the medical staff. This will ensure the highest probability of adoption of the product. The importance of including transit and application issues can be shown with an example. Within the Directive the definition of an ‘industrial process’ is unclear. There is uncertainty about where manufacture ends and the ‘practice of medicine’ begins. An example can be seen in the technology of bioprinting. A cell-and-scaffold construct can be printed to make a customised implant. Such a product may be made close to the clinic and may require trimming by a surgeon in theater before implantation. The trimming may be regarded as a part-finishing activity in the manufacturing process or as a part of the surgical procedure.
Advantages of Centralization
When manufacturing began most goods were made locally. During the industrial revolution economies of scale were realized in the large factory. The benefits of centralization are a) spreading the costs of doing business over large production volumes (minimization of overheads), b) reducing interactions with key suppliers and key customers and c) smoothing the impact of local peaks and troughs in demand by making a variety of products re-deploying resources rather than retaining idle resource. Centralization has disadvantages as well. A single location can make it difficult to access distant markets. Responsiveness of manufacturing may not be as high as it could be if it were close to the point of consumption.
As described earlier, the results of analysis are a necessary but insufficient condition for assurance of quality. Other evidence must come from the demonstration of compliance of the manufacturer with the registered conditions of manufacture. Such compliance is monitored in the EU by the Qualified Person or in the US by the manager of the Quality Unit including inspection of practice and of batch records. Under GMP there is a requirement to report on any potentially significant trends in manufacture, preferably before the batches concerned have begun to fail the tests for market release. A cautious strategy, and the easiest to implement, is to carefully design a manufacturing process for a single site and then to maintain this for the product lifetime.
Requirements of CBTs
CBTs have characteristics that influence the choice of manufacturing, distribution and delivery. These can be regarded as drivers of choice. The drivers can be classified for convenience as quality drivers, cost drivers or supply drivers.
Minimizing supply chains, maximizing market penetration (quality, supply & cost drivers)
Long-distance transport of goods can be expensive; in particular as CBTs need careful control of temperature and vibration during transit. Anecdotally the impact on ex-factory cost for allogeneic engineered tissue is expected to be an increase of approximately 50% for global low temperature using non-returnable dry shippers. This cost may be sustained for high-value, low-volume therapies. The cost becomes untenable as margins decrease and volumes increase. Some therapies are best supplied ‘fresh-preserved’ i.e. at 37°C (for example some chimeric antigen receptor (CAR)-T therapies [13]). Figure 1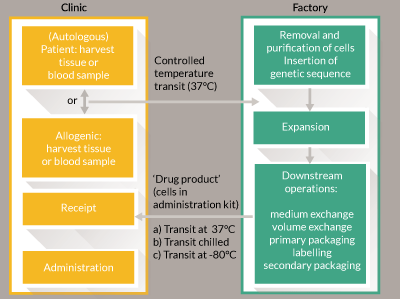
Minimizing warehoused material (supply & cost drivers)
The reduction of goods held in stock is to reduce cost of operation. Excess or large stock holding is a liability because it depreciates in value as a perishable good and requires outlay to acquire and to maintain. This cost is minimized if the goods are made to order (MTO). MTO manufacture is easier to achieve for autologous products for non-critical indications. Such a supply arrangement requires good responsiveness and robust procurement and order procedures. The responsiveness is easier to achieve over short supply distances and is a driver towards distributed manufacture.
Terminal customization (supply & quality drivers)
A high proportion of cell therapy products are simple injectable products. Products made for direct injection into easily accessible sites may be applied using simple or slightly modified syringes for their application. However, there is a product category of growing importance in which a combination of applicator and cells is an advantage [15]. The medical device component of a combination product is generally stable to storage in undemanding conditions for long periods. The device manufacture will be subject to control according to a quality management system such as ISO 13485, which imposes a smaller overhead burden than does manufacture to GMP. The cellular component will require much higher levels of manufacturing and environmental control during storage and will be subject to GMP. The combination of the two can be manufactured either in a fully GMP-compliant facility or in one that contains appropriate safeguards from the point at which the two components are combined. This means that there is an operational advantage in being able to bring the two components together at as late a stage in production as possible for an area-dependent dosage form (e.g., a cellular patch for cartilage repair) or a volume-dependent dosage form (e.g., bone graft substitute material containing cells). There is an advantage in postponing the introduction of cells to allow custom features to be included. This situation is well illustrated in bioprinting of implants where the goods are made by printing cells and materials concurrently. The activity is best carried out close to clinic.
Economy of scope (cost driver)
The contribution of the cost of the manufacturing facility to each unit manufactured is found by dividing the write-off cost plus ongoing maintenance cost by the number of units made during the interval under consideration. Bespoke equipment can be expensive to hold and to maintain. For a market where fluctuations in demand occur, the equipment will be subject to down-time when demand is low. The costs of the facility can be reduced by maximizing the use of the resource for production. In a centralized facility this is achieved mainly by the familiar practice of operating fixed shifts to manufacture around the clock. The strategy is most effective for a centralized facility because the market catchment area is large and local variations average out. An alternative strategy, more familiar to contract manufacturing organizations (CMOs), is to apply economy of scope. To do this the manufacturing equipment must be capable of producing more than one product. The cost is spread across several products and equipment utilization is higher. For centralized operation this strategy is feasible and, indeed, established through familiar production, for example at Lonza (Maryland, USA), Cognate (Maryland, USA) and Progenitor Cell Therapies (New Jersey, USA), Inc. Economy of scope in a distributed network of manufacturing requires a common manufacturing platform. This may be a stirred-tank or Wave® bioreactor (GE Healthcare, Pennsylvania, USA), but dedicated systems that have been designed for specific products do not offer this potential and may drive manufacturing costs up for small operations.
Control of recalls (quality driver)
An important responsibility for a manufacturer of medicinal products is to be able to rapidly and effectively recall defective goods. A robust track-and-trace capability is needed as part of Corrective And Preventative Action (CAPA) under GMP. The oversight and authority to act swiftly and effectively will be more easily achieved in a centralized manufacturing site.
Management of retained stock (cost & quality driver)
It is common practice to retain a part of the batch in case adverse behavior is found in a marketed product and it becomes necessary to examine the batch more thoroughly. For a CBT the sampling, secure storage and record-keeping is a significant cost burden. The burden increases as the number of batches increases and the size of each batch decreases. This is an area where the economies of centralization are evident. If a product is to be made and supplied fresh- or chilled-preserved (4–8°C) then the fact that the archive must be frozen at low temperature in order to store it means that the archive and the administered product differ and retained stock may be an irrelevance.
Capacity & market growth (supply & cost drivers)
When a product is launched it is essential to meet demand in order to retain the confidence of customers, especially if there is a choice of treatment available. This means building adequate manufacturing capacity at the outset in order to avoid capacity shortfalls while building and validating new plant. A distributed manufacturing model may permit the construction of regional facilities in line with market expansion into those regions. The capital put at risk is thus minimized. However, this risk reduction can only be realized if the challenge of comparability is overcome.
Disaster recovery (supply driver)
If the manufacturing facility is damaged, for example by fire, natural disaster or infection, supply will be interrupted, perhaps permanently. Centralized manufacturing is vulnerable to this threat; distributed manufacturing less so. In a distributed manufacturing network it may be possible for staff and resources to be redeployed between sites in order to maintain supply.
Business models
In order to choose between centralization and decentralization and their possible variants we must have a tool to examine the advantages and disadvantages of each: we need business models [16]. The way of thinking about the models of business that may be used for the manufacture of cell therapies has undergone several changes in the approximately 50 years that the sector has been growing [17].
The term business model is used to define the means by which a value proposition [18] may be converted into a revenue stream. The study of business models began to accelerate in the 1990s [19] at about the same time as the first companies began to market CBTs. There has been debate over the usefulness of business models [20] but they remain one of the very few tools that can be used to articulate and to evaluate options for generating revenue. Given that it is expensive to make late-stage changes to the manufacturing method, it is prudent to have a vision, preferably a quantitative one, of the costs of operation as early as possible. A good business model is a management device that is used to manage the sometimes conflicting factors in the business environment [20]. It must be used to explore a market and to translate the innovation into wealth generation. It defines the position of the enterprise in a value chain. A good model is explicative and predictive.
CBTs may be classified at the highest level as allogeneic or autologous. More companies are addressing autologous therapies as technology ‘push’ coupled with market demand are making personalized medicine more attractive. Intra-operative cell-based therapies are increasing. There is growth in delivery of CBTs based upon a product status of ‘hospital exemption’ [5] or ‘special’. Many companies entering the market for CBTs are start-ups and small and medium-sized enterprises (SMEs). It is important for such entrepreneurs to appreciate quickly the factors that may influence their choice of business models.
To illustrate the balance of influences on the decision to centralize or to decentralize a good example is CAR-T cell therapies. Consider a hypothetical business in which the current research is at the preclinical stage. The product is ‘personalized’: although both autologous and allogeneic therapies are in development they resemble autologous therapy manufacture because it is not practical to work from a single cell bank. Instead the product is derived for each patient either from their own cells via an apheresis procedure or from the cells of a related donor. They are closely equivalent because each requires a scaled-out process with an upstream cell harvesting step, a manufacturing step that includes genetic modification and expansion and a downstream transport and administration step. In this example we have a high-value product because the condition (typically acute leukemia) is likely to be fatal if untreated. So far the success rate has been very high [13]. Unlike the previous example, it is possible to justify lower-volume, more labor-intensive manufacturing, with operational costs absorbed in the overheads. When supplying there is a pressing need to coordinate product release with delivery to clinic. Release is based upon the results of quality control testing and the duration of this may be too long for fresh-preserved goods to be held pending authorization. The options for this step are shown in Figure 2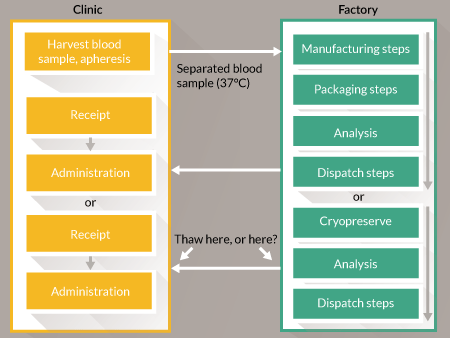
The technical challenge is to gather sufficient evidence to release the product ‘at risk’ before all the results become available. A similar situation exists for three product categories: radiopharmaceuticals, parenteral nutrition products and some chemotherapeutic agents that are formulated in patient-specific doses. In those categories the products are well-defined and, as noted above, the burden of proof for assurance of quality will not be as heavy. CAR-T cell therapies are more complex. Some must be delivered and used as quickly as possible after manufacture. The solution here appears to be to choose from one of the three options below and to generate evidence to justify the choice:
- Cryopreserve the goods following manufacture and hold pending full testing. (Early research must prove that the cryopreserved product will be as safe and efficacious as the fresh product.)
- Release the goods ‘at risk’ from a well-controlled process. (Early research must provide evidence of high process capability.)
- Move the assurance of quality to real-time measurements during manufacture so that the data are available immediately at point of release.
In option 3 the basis of measurement may be either a) in-line or at-line measurements (real-time release technology [21]), or b) the correlation of the behavior of manufactured batch with the behavior of satisfactory calibration batches. The latter is only really practical if feedback control is applied; open-loop control methods provide no evidence of process response.
Fresh preservation favors a decentralized business model in order to minimize supply time and distance. Novartis (Basel, Switzerland) is an important innovator of CAR-T therapies. The Novartis/University of Pennsylvania research and training facility at Morris Plains has many of the attributes of a ‘franchise prototype’. A franchise prototype is a facility with equipment, training scheme, quality management system and process manufacturing instructions that can be transferred directly to another manufacturing site of similar size, for example in or near a hospital. This strategy can potentially be very capital-efficient if the business introduces such new centers incrementally. (See ‘Scaling in line with market growth’ below.) To achieve this, the challenge of comparability arises in a different way. Bridging studies become necessary to demonstrate that manufacture in different parallel sites and with different teams can produce equivalent product. Alongside this is another hurdle related to registered facilities and legal liability. Whether the US or the EU system is considered (registered manufacturing sites subject to FDA inspection or Manufacturing Authorisations issued to named facilities under Site Master Files) it is normal to register one or two centers only. To manufacture from many centers is to pose the problem of what the legal entity is that is accepting liability for product quality and how many separate licenses are required to secure public safety. Three basic formats of operation can be envisaged. Not all of the variants are economically viable:
- The distributed factory
- The franchised business
- The autonomous microfactory
The distributed factory
One legal entity with a single license to manufacture bears responsibility for every location where manufacture takes place. The legal entity, probably a large biopharma business (in order to provide the required scale, reach and consistency of support services) provides staff, equipment, quality management system and consumables to each manufacturing location. Each location is subject to management control from the center. Internal inspections, purchasing, supplier audits, quality management and regulatory affairs services are carried out from the main business hub. In order to achieve these criteria it is important to allow for compliance to GMP across the regions in which the distributed factory will operate. An example of this planning is seen in the facility at the Lonza, Walkersville operation in the USA which contains four suites constructed and operated to EU Grade B cleanroom background and regulatory compliance. A UK example of such practice exists in the operational management of the NHS Blood and Transplant cleanrooms which manage the production protocols to common standards across multiple sites.
The franchised business
This resembles the ‘distributed factory’ but there are important differences. The description that follows is speculative because there are, to the best knowledge of the author, no current examples of this model in operation. The central business offers the franchise based upon a package of data similar to that which is currently generated for the chemistry, manufacturing and controls package in the marketing authorisation and for the Site Master File. The franchisor assists the franchisee in their project to gain a Manufacturing Authorisation for their production site. Operations at the franchisee site are conducted to a boilerplate quality management system built by the franchisor. Each new manufacturing operation bears legal liability for assurance of quality from the point of view of production management and operations management. The business that offers the franchise (equivalent to the hub in the previous example) bears responsibility (via the QP or quality unit oversight, depending on territory) for release of goods. The franchise-holder organization is only permitted to operate if it remains in compliance with the franchise terms and conditions. Compliance will be subject to periodic audit and verification. A technical refinement to this model, operable only in the case of the ‘autonomous microfactory’ (see below), is to increase control of quality by the franchisor by retaining authority for release of goods. That authority would be managed remotely based on data transmitted from the franchise holder to the franchisor. The data would be subject to electronic scrutiny under human oversight before authorising release of goods.
The autonomous microfactory
Process automation is the best method for ensuring that a manufacturing process is carried out reproducibly to the same control strategy at different times and in different locations [22]. The autonomous microfactory is a self-contained machine that operates to a firmware program containing a specified set of process instructions. Each process instruction operates the microfactory to make one product. Operator discretion is reduced to a minimum. The microfactory records the measurements made during manufacture and collates them as part of the batch records. There are several extant examples of the microfactory currently in development and these are described in Table 1.
| Research group or company | Technology | Key features |
|---|---|---|
| Aastrom Biosciences, Inc. | Aastrom Replicell System (ARS) | Closed parallel plate bioreactor |
| Automated protocols for cell washing and expansion | ||
| Clinical-grade product | ||
| Kawasaki Heavy Industries Ltd. with National Institute of Advanced Industrial Science and Technology (AIST, Japan) | R-CPX: Robotized - Cell Processing eXpert system | Robots mimic the work of technicians |
| Image processing for cell assessment | ||
| Cell history management | ||
| Flexible production scheduling | ||
| Octane Biotech, Inc. | Octane Cocoon | Pod-like closed manufacturing unit |
| Internal disposable wetted components | ||
| Autologous products made in clearly-separated units with independent control | ||
| Osaka University (Prof. Kino-Oka, Masahiro) | Kotozukuri (construction of a industrial system to connect all the essential processes) flexible Modular Platform (fMP) | Normalisation of fluidics, handling and transfers |
| Self-contained microfactory system | ||
| Manages its own atmospheric environment through proprietary interlocks | ||
| Re-configuration of isolators, permits different production layouts from single microfactory | ||
| TAP Biosystems | CompacT Select® | T-flask handling |
| Multiple cell line handling | ||
| Validatable | ||
| Plating options | ||
| Reproducible liquid handling | ||
| Capacity options | ||
| Tokyo Electron Limited (TEL) | Fully-automated Smart Cell Factory | Colony isolation with automated cell picking and culture |
| Standardised bespoke culture plates to normalise colony conditions | ||
| Clinical grade output |
An established example of the closed microfactory concept in current use is the CliniMACS Prodigy®(Miltenyi Biotec, Germany); a GMP-compliant system. Optical sensor measurements identify layers of plasma and cells collected from a mini-centrifuge. Cell culture is carried out in a closed chamber and user variability is largely eliminated by the controls. The system is complete with heat sealing apparatus for aseptic tubing connections and is controlled by customizable process software. It was developed as a medical device and manufactured under ISO 13485 compliance but its products are made under Investigational New Drug or Investigational Device Exemption criteria. The CliniMACS Prodigy® is currently indicated for treatment of acute myeloid leukemia in patients receiving CD34+ allogeneic cell transplants from immunologically-matched donors.
The removal of the risk factors and the normalization of expectations for product quality might be expected to be a persuasive and final consideration for manufacturing where distribution is advised. In practice there are two more major considerations that must be made before such technology may be rolled out as a universal platform. The first of these is the issue of capital efficiency and the second is the management of trends in microfactory performance.
Capital efficiency & microfactories
Requests for heavy investment in unique capital equipment have never been popular with investors. Since the 2008 downturn venture capitalists have become less tolerant of proposals for high investment in bespoke capital equipment [23]. This is because, in the event of a business failure, the resale value of such equipment will be negligible. ‘Capital efficient’ investments are favored i.e., ones that avoid large outlay at risk on new equipment as much as possible. There are three main ways of remaining capital efficient: to use CMOs; to remain in academia as long as possible; or to increase capacity by scaling out, rather than scaling up, using largely manual processing. The first two choices carry the risk that existing plant proves unsuitable. The third is, as we have seen, prone to variation and to risk of non-compliance.
Examples of CMOs working well for CBT businesses include Tigenix (hosted by Cognate), Mesoblast (Lonza) and translational centers such as the California Center for Regenerative Medicine, Progenitor Cell Therapies and the Cell and Gene Therapy Catapult. The use of a CMO requires either that the unit operations will fit into existing technology platforms or that bespoke equipment, such as the autonomous microfactory, can be accommodated economically.
Autonomous microfactories are expensive to acquire and to validate. For high-price, high-value products the maintenance costs and write-off period will be absorbed by the product margins. For mid-price products they are not so economically attractive. In order for them to become viable it will be necessary to spread the cost of operation across several products, thus maximizing use of the asset as described above. It is unusual for a single company to launch several cell products at the same time. A versatile platform technology could be used by several companies to launch products with similar manufacturing requirements over a short interval. To enable this to happen the platform technology would need to be made to a specification which has been shown to be needed for a range of products with common characteristics. This is likely to happen only if several potential users act together to signal the specification to equipment producers. The business confidence needed before an automation provider will commit to developing suitable technology is unlikely to come from one innovation alone. The confidence to establish common platforms serving more than one product type must come from collective action within the industry. There is an important opportunity to be explored by industry bodies and standards agencies, such as the Medicines Manufacturing Industry Partnership, the International Standards Organisation and the European Federation of Pharmaceutical Industries and Associations in bringing the user community together at a pre-competitive level. If such platform technologies were available they would facilitate technology transfer, they could be applied early in research (reducing or removing much of the development stage) and, most importantly, they could be established as the workhorse of manufacturing hubs operating as translational facilities for several innovating companies at the same time.
Management of trends in microfactory performance
GMP requires that cell therapy businesses monitor and respond to observed trends in the quality of their manufactured goods [24]. In a centralized business this task is straightforward and the results are considered during Quality Reviews. In a distributed manufacturing operation the task is more demanding because each microfactory will be further away from the central Quality Unit and there will be fewer opportunities for direct oversight and routine observation. There will be a higher risk of drift away from calibration in each microfactory. The risk can be reduced if specified sets of starting materials and consumables are generated for regular calibration purposes. During a manufacturing campaign these could be introduced into the system, either during periodic re-calibration runs or ‘blindly’ by introducing dummy orders based on positive and negative control material, in order to test the performance of the microfactory periodically. This is currently an ambitious plan because it means correlating the important features of these materials with known manufacturing outcomes.
Scaling in line with market growth
Scaling out on a decentralized manufacturing model can provide a significant advantage when there is uncertainty about the rate and volume of market penetration. The aim is to build in line with market demand. This phased introduction results in a gradual increase in market revenue but commits capital investment only in line with growth, thus limiting the downside of the investment. The approach relies on one of two alternatives: establishing a central facility and increasing capacity from that location or the generation of franchise-style parallel operations at different locations. Each option has its strengths and weaknesses. This would need to be justified in practice by a sufficiently high product value and the ability to maintain competitive position long enough to manage the lead times to bring successive manufacturing units on stream. The scenario is easier to justify if common technology platforms have already been established across CMOs in different territories.
TRANSLATIONAL INSIGHT
The choice of business models for CBTs is best made early in the product development cycle. This avoids costly repeat work to ensure that any late-stage process changes give the product comparable performance to the material assessed in the pre-clinical studies. Distributed models of manufacture offer some attractive advantages in terms of cost and flexibility. The challenge of proving that the same quality of product is made site-to-site remains considerable. Automation may provide the key to this challenge but remains expensive. Spreading the cost of capital equipment by providing economy of scope requires industry standard manufacturing platforms. Such standards are most likely to arise through collective action by the industry and research community and cannot be left to individual companies. A pre-competitive community of practice is required to bring users and providers to shared conclusions about the technical solutions that are needed. With the CBT market poised for growth, the success of new businesses and the degree of patient access to new products will be affected in large measure by the degree of ‘fit’ of the manufacturing and supply operations to the financial, quality and cost drivers for the sector.
In the UK a systems-level approach to manufacturing and supply is recognized as likely to accelerate translation [25]. The successful factory of the future will involve organizational and technical innovations. Agile businesses that use appropriate supply chains and chains of custody will be the most successful [26].
Financial & competing interests disclosure
The author has no relevant financial involvement with an organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock options or ownership, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Disclaimer
While the contents of this review are, to the best knowledge of the author, accurate at the time of publication no liability can be accepted for decisions based upon the content. Readers are advised to consult Investment, Quality Assurance and Regulatory professionals concerning decisions about their own business practice.
References
1. Hampson B, Rowley J, Venturi N. Manufacturing patient-specific cell therapy products. BioProcess Int. 2008; 60–72.
2. Medcalf N, Hourd P, Chandra A, Williams DJ. Quality assurance and GMP in the manufacture of cell-based therapeutics. StemBook (ed.) 2014; The Stem Cell Research Community, StemBook, CrossRef
3. European Commission. Regulation (EC) No 1394/2007 of the European Parliament and of the Council of 13 November 2007 on advanced therapy medicinal products and amending Directive 2001/83/EC and Regulation (EC) No. 726/2004. Official Journal of the European Union. 2007; L 324/121.
4. European Commission. Directive 2011/83/EC of the European Parliament and of the Council of 6 November 2001 Relating to Medicinal Products for Human Use. Official Journal of the European Union. 2004; 311, 67–128.
5. Cuende N, Boniface C, Bravery C et al. The puzzling situation of hospital exemption for advanced therapy medicinal products in Europe and stakeholders’ concerns. Cytotherapy 2014; 16, 1597–1600. CrossRef
6. Salmikangas P, Celis P. Current challenges in the development of novel cell-based medicinal products. Regulatory Rapporteur 2011; 8(7/8), 4–7.
7. a) European Medicines Agency. ICH Q6A: Specifications – Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances. 2000. www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002823.pdf.
7. b) European Medicines Agency. ICH Q6B: Specifications – Test Procedures and Acceptance Criteria for Biotechnological/Biological Products. 1999. www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002824.pdf.
8. Salmikangas P, Menezes- Ferreira M, Reischl I et al. Manufacturing, characterisation and control of cell-based medicinal products: Challenging paradigms toward commercial use. Regen. Med. 2015; 10(1), 65–78. CrossRef
9. Hourd P, Chandra A, Medcalf N, Williams DJ. Regulatory challenges for the manufacture and scale-out of autologous cell therapies. StemBook, (ed.) 2014; The Stem Cell Research Community, StemBook, CrossRef
10. Schlegel M & Bobinnec Y. Comparability protocols for biotechnological products. BioProcess Intl. 2013; 11(6), 30–41. CrossRef
11. Bravery C. Comparability: Managing Process Change. Conference: Challenges toward sound scientific regulations of cell therapy products. International Alliance for Biological Standardization. Kyoto. 2014; 7–8 March.
12. Medcalf N. A new business model for cell-based therapeutics. 2011; PhD thesis, NUI Galway.
13. Paulson W (editor). Novartis Exploring Boundaries of Biotech Manufacturing and Control to Make Cell and Gene Therapy Commercialization a Reality. International Pharmaceutical Quality. 2015; 6(2), 3–12. www.ipqpubs.com/wp-content/uploads/2015/05/IPQ-Monthly-Update-March-2015.pdf
14. Nordström KM, Närhi MO, Vepsäläinen APJ. Services for distribution of tissue engineering products and therapies. Int. J. Prod. Perf. Manag. 2009; 58(1), 11–28. CrossRef
15. Lyness A, Williams DJ, Medcalf N. Overcoming the translational challenges of the effective administration and delivery of cells. Cytotherapy 2015; 17, S83–4 CrossRef
16. Omidvar O, De Grijs M, Castle D, Mittra J, Rosiello A, Tait J. Regenerative medicine: Business models, venture capital and the funding gap. 2014; Innogen and ESRC report. http://www.innogen.ac.uk/reports/904.
17. Brindley DA, Davie NL, Sahlman WA et al. Promising Growth and Investment in the Cell Therapy Industry during the First Quarter of 2012. Cell Stem Cell 2012; 10, 492–6. CrossRef
18. Boons F, Lüdeke-Freund F. Business models for sustainable innovation: state-of-the-art and steps towards a research agenda. J. Clean. Prod. 2013; 45, 9–19. CrossRef
19. Zott C, Amit R, Massa L. The Business Model: Recent Developments and Future Research. J. Manag. 2011; 37(4), 1019–42. CrossRef
20. Doganova L, Eyquem-Renault M. What do business models do? Innovation devices in technology entrepreneurship. Res. Policy. 2009; 38, 1559–70. CrossRef
21. Kirouac DC, Zandstra PW. The systematic production of cells for cell therapies. Cell Stem Cell 2008; 3, 369–81. CrossRef
22. Brindley DA, Wall IB, Bure KE. Automation of cell therapy biomanufacturing. BioProcess Intl. 2013; 11(3), 18–25. CrossRef
23. Bonfiglio GA. Why Invest In RM? World SCRM Congress 2015 – Investor Forum – Horizon Scoping (London May 22, 2015)
24. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonized Tripartite Guideline: Good manufacturing practice guide for active pharmaceutical ingredients, Q7. 2000. www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q7/Step4/Q7_Guideline.pdf.
25. Regenerative Medicine Expert Group. Building on our own potential: a UK pathway for regenerative medicine 2015; www.gov.uk/government/publications/regenerative-medicine-a-uk-pathway.
26. Ridgway K, Clegg CW, Williams DJ. The factory of the future: A study for the Government Office for Science. The National Metals Technology Centre, University of Sheffield AMRC 2013.
Affiliation
Nicholas Medcalf
EPSRC Centre for Innovative Manufacturing in Regenerative Medicine,
Centre for Biological Engineering,
Garendon Wing,
Holywell Park,
Loughborough University,
Loughborough, LE11 3TU, UK
n.medcalf@lboro.ac.uk