MSC homing & immunomodulatory properties in cancer therapies: searching for the perfect balance
Cell Gene Therapy Insights 2015; 1(2), 173-192.
10.18609/cgti.2015.024
Mesenchymal stem cells are non-hematopoietic adult stem cells with multi-lineage potential. Their inherent tumor tropism and easy isolation, expansion and transduction, make them attractive vehicles for the delivery of anti-cancer agents. Mesenchymal stem cell tumor homing is still poorly understood and a wide variety of factors have been reported to affect this complex process, with some inconsistencies. Their immunomodulatory properties have led to some caution towards their use in cancer patients but this field remains controversial, as both immunosuppressive and immune-enhancing phenotypes have been described and appear reversible as well as highly sensitive to the local microenvironment. This review will focus on mesenchymal stem cell homing and immunobiology in the context of cancer and the translational potential of these cells.
Submitted for Review: Oct 15 2015 Published: Dec 10 2015
This review will discuss the recent developments in several important areas of mesenchymal stem cell (MSC) biology, although it is difficult to draw definitive conclusions; this is largely due to the plethora of cell sources and culture conditions used in this field and the subsequent heterogeneity in cell phenotype denoted by the term MSC. To address this, the International Society for Cellular Therapy have proposed minimum criteria for application of the term ‘MSC’:
- Adherence to tissue culture plastic;
- Absent expression of CD45, CD34, CD14, CD11b, CD19 and HLA-DR;
- Cell surface expression of CD105, CD73 and CD90; and finally
- Potential for in vivo differentiation to osteoblasts, adipocytes and chondroblasts under standard conditions [1].
However, a revision of these criteria may soon be required for example taking into account the distinction between high and low growth capacity MSCs [2].
Homing to tumors
Extravasation
MSC homing to tumors is thought to be due to inflammatory signalling in a tumor resembling that of an unresolved wound [3]. The mechanism and key players responsible for this tumor-targeted tropism remains to be fully elucidated. It is hypothesized that MSCs behave similarly to leukocytes and have chemotactic properties allowing response to a variety of secreted chemokines from tumor cells. Several studies have correlated an increase in circulating MSCs with increases in inflammatory cytokines [4–6]. Despite some key observations, the initial steps in MSC mobilization and intravasation into the blood stream remain unclarified.
To understand MSC homing, many investigators have studied well known factors involved in leukocyte homing as a starting point for investigation. Leukocyte chemotaxis is a multistep process involving capture, rolling, activation, arrest, adhesion, crawling, transendothelial migration and engraftment. Selectins, selectin ligands, integrins-immunoglobulin superfamily receptors (vascular cell adhesion molecule-1[VCAM-1]), chemokines and their receptors, and a variety of other molecules play important roles in this process [7]. While there are as yet unresolved differences in studies it would appear MSC homing may use similar molecules and cell–chemokine interactions to leukocyte trafficking (reviewed in [8]).
The first step in recruitment and engraftment of MSCs is the exit from the vascular circulation (extravasation), which requires crossing the blood vessel endothelial cell (EC) barrier. For MSCs, this process of extravasation remains unclarified [9]. Leukocyte extravasation at sites of inflammation has been studied in depth and is characterized as a rapid multistep cascade. Within an inflammation context, the endothelium becomes activated by cytokines such as tumor necrosis factor (TNF)-α. Chemoattractants and surface proteins, including selectins and cell adhesion molecules (CAMs) are upregulated as a consequence, mediating rolling and adhesive interactions, respectively (Figure 1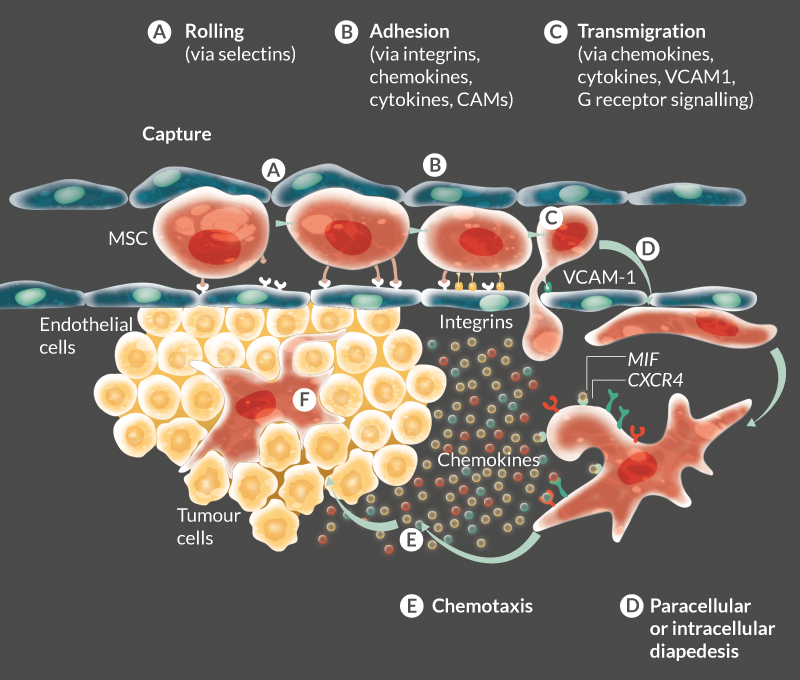
Very few studies have investigated MSC transmigration. The data generated from these studies led to the hypothesis that MSCs incorporate the endothelial monolayer following the retraction of ECs [13–15]. However, the molecular and cellular details of this process such as detailed 3D cellular architecture, distribution of adhesion, endothelial junction molecules, and interactions have not yet been investigated and remain unclarified. Teo et al elegantly used high-resolution confocal and dynamic live-cell imaging to show that MSCs can transmigrate in a leukocyte-like way by paracellular and transcellular diapedesis, in association with endothelial transmigratory cups [9]. However, they did not observe MSC transmigration by the formation of lamellipodia or invadosomes – instead, MSCs formed membrane blebs allowing endothelial crossing. The process was also significantly slower than that seen in leukocytes and was suggested to occur via VCAM-1 and G receptor signalling-dependent mechanisms [9]. Several studies have suggested that MSC homing to tumors occurs by a combination of active recruitment by chemokines and inflammatory processes as well as passive entrapment in the vasculature due to their size (Figure 1).
Chemotaxis
Multiple adhesion molecules, integrins and chemoattractants play established roles in leukocyte trafficking and may have significant overlap with MSC homing mechanisms [16]. A variety of different ligand/receptor pairs have been identified as players in the process, as shown by Table 1 listing markers and receptors typically associated with cell migration and known to be expressed in MSCs (reviewed in [3]).
| Table 1. Cell surface markers expressed on MSCs associated with cell migration/homing and their respective ligands. | ||
|---|---|---|
| Cell surface receptors found on MSCs | Ligands | |
| Growth factor receptors | EGFR | EGF |
| HGFR | HGF | |
| IGF1R | IGF1 | |
| PDGFR | PDGF | |
| VEGFR1 | VEGF | |
| VEGFR2 | VEGF | |
| FGFR2 | FGF2 | |
| Tie-2 | Ang-1 | |
| TGFβRI | TGFβ1/2 | |
| TGFβRII | TGFβ1/2 | |
| Chemokine receptors | CCR1 | CCL3, CCL5, CCL7, CCL13, CCL14, CCL15, CCL16, CCL23 |
| CCR2 | CCL2, CCL7, CCL8, CCL13, CCL16 | |
| CCR3 | CCL5, CCL7, CCL8, CCL11, CCL13, CCL15, CCL16, CCL24, CCL26, CCL28 | |
| CCR4 | CCL17, CCL22 | |
| CCR5 | CCL3, CCL4, CCL5, CCL8, CCL11, CCL14, CCL16 | |
| CCR6 | CCL20 | |
| CCR7 | CCL19, CCL21 | |
| CCR8 | CCL1 | |
| CCR9 | CCL25 | |
| CCR10 | CCL27, CCL28 | |
| CXCR1 | CXCL6, CXCL7, CXCL8 | |
| CXCR2 | CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7, CXCL8, MIF | |
| CXCR3 A/B | CXCL4, CXCL9, CXCL10, CXCL11 | |
| CXCR4 | CXCL12, MIF | |
| CXCR5 | CXCL13 | |
| CXCR6 | CXCL16 | |
| CX3CR1 | CX3CL1 | |
| XCR1 | XCL1, XCL2 | |
| Cytokine receptors | IL1R | IL1α, IL1β IL1RA |
| IL3R | IL3 | |
| IL4R | IL4, IL13 | |
| IL6R | IL6 | |
| IL7R | IL7 | |
| IFNγR | IFNγ | |
| TNFRI and II | TNFα, TRAF2, TRAAD | |
| Cell–matrix receptors | CD44 | Hyaluronan |
| α1β1 (VLA-1) | Collagens, laminins | |
| α2β1 (VLA-2) | Collagens, laminins | |
| α3β1 (VLA-3) | Laminin-5 | |
| α5β1 (VLA-5) | Fibronectin, proteases | |
| α6β1 (VLA-6) | Laminins | |
| αvβ1 | Vitronectin, fibrinogen | |
| αvβ3 (vitronectin receptor) | Vitronectin, fibronectin, fibrinogen, ostopontin, Cyr61 | |
| α1β5 | Vitronectin | |
| Cell–cell receptors | VCAM-1 | β1 integrin / α4 integrin |
| ICAM-1/3 | LFA-1 | |
| ALCAM | CD6 | |
| CD105 | TGFβ1/3 | |
| Immuno-modulating receptors | TLR1 | Lipopeptides, peptidoglycan |
| TLR2 | Peptidoglycans, lipopeptides | |
| TLR3 | dsRNA | |
| TLR4 | LPS | |
| TLR5 | Flagellin | |
| TLR6 | Peptidoglycans, lipopeptides | |
| TLR9 | Unmethylated CpG DNA | |
| Adapted from Spaeth et al [3]; dsRNA: double-stranded RNA; EGF: epidermal growth factor; EGFR: epidermal growth factor receptor; FGF: fibroblast growth factor; FGFR: fibroblast growth factor receptor; HGF: hepatocyte growth factor; HGFR: hepatocyte growth factor receptor; IGF: insulin-like growth factor; IGFR: insulin-like growth factor receptor; IL: interleukin; LFA: lymphocyte function-associated antigen; PDGF: platelet-derived growth factor; PDGFR: platelet-derived growth factor receptor; LPS: lipopolysaccharide; TGFbRI: transforming growth factor beta receptor I; IFNγ: interferon gamma; TNFα: tumor necrosis factor alpha; TRAF: TNF receptor-associated factor; TLR: toll-like receptor; VEGF: vascular endothelial growth factor; VEGFR: vascular endothelial growth factor receptor. | ||
The most extensively studied MSC chemotactic axis is CXCR4/ stromal cell-derived factor (SDF)-1. High levels of SDF-1 have been shown to be important in recruiting and retaining hematopoietic stem cells (HSCs) within bone marrow [17], and there is increasing evidence that cancer cells home to bone marrow following similar CXCR4 expression [18–21]. Soluble chemoattractants secreted by tumor cells have been suggested to activate MSC migration by triggering them to secrete SDF-1. Recently, investigators found that soluble factors secreted from tumor cells can trigger SDF-1 secretion from MSCs, activating their migration [22]. The role of SDF-1 is disputed, however, as several studies have shown that tumors generally do not produce SDF-1 [23].
We recently identified macrophage migration inhibitory factor (MIF) as a key factor through screening soluble factors secreted from tumor cell lines [24]. Other studies have shown MSC migration to be regulated by numerous other factors including tumor cell-specific receptors, extracellular matrix and soluble tumor-derived factors such as SDF-1, TNF-α and interleukins (ILs) [22,25]. MSCs are undoubtedly responsive to other chemokines. In our studies, we also see stimulation of MSC migration towards gradients of IL1β, IL6, IL8 and CCL2. Roles for IL6, IL8 and CCL2 have been delineated by several other groups [26–29]. Similar to the work on SDF-1, we see up-regulation of IL1β, IL6, IL8 and CCL2 in MSCs treated with tumor-conditioned medium. Interestingly we observe chemotactic effects with these four cytokines, suggesting a positive feedback loop possibly leading to amplification of chemoattraction to tumor cells, likely triggered by MIF. This MIF-dependent amplification is seen in other studies showing MIF-dependent up-regulation of these cytokines in an inflammatory context [30–33].
We have confirmed recombinant SDF-1 as a chemoattractant for MSCs. However we do not believe it has a role in the in vivo tumor context after failing to detect significant levels secreted by several cancer cell lines (A549, MDAMB231, H376, A431 or Jurkat cells) and a failure to block migration of MSCs to these tumor cells with an SDF-1 neutralizing antibody. Of note, MSCs did show reduced migration with SDF-1 blockade towards the U87MG cell line (described to secrete SDF-1 [34,35]); however even in this cell line, migration was more severely diminished by CXCR4 or MIF antagonists [24].
These results reinforce our hypothesis that MIF is the dominant chemoattractant in the recruitment of MSCs to tumors, even in the presence of SDF-1 (Figure 1). These findings are consistent with other reports showing high secretion levels of MIF and rare secretion of SDF-1 in the majority of tumors [23,36–42]. MIF–CXCR4 has been described as important in a variety of other contexts: regulation of endothelial progenitor cell migration, cancer metastasis and cancer proliferation/growth [43].
MSC immunogenicity
MSCs have long been described as hypoimmunogenic or immune privileged, and this property has been widely explored for the creation of ‘off-the-shelf’ MSC therapies as a means of circumventing major histocompatibility barriers. However, several preclinical and clinical observations have led to controversy as to the true presence and extent of MSC immune privilege and their subsequent potential for universal donor therapies (reviewed in [44]). Questioning the immune privileged status of MSCs, an elegant study by Zangi et al compares the persistence of syngeneic and allogeneic MSCs in vivo [45]. The authors show a significant decline in detectable MSCs in an allogeneic setting. Others have also shown innate immune responses to allogeneic MSCs [46,47]. Disparities between reported results may be due to discrepancies between MSC microenvironments. Culture-expanded MSCs express low levels of major histocompatibility complex (MHC) class I antigens, and are negative for MHC class II; however, following treatment with IFN-γ, MSCs have been shown to up-regulate these markers [48]. The timing and severity of MSC rejection appears to be strongly dependent on context and dictated by a balance between the expression of immunogenic and immunosuppressive factors. Intriguingly, when the immunosuppressive phenotype is not activated, MSCs may acquire an antigen presenting-like phenotype, able to promote an immune response in vivo [49,50]. Harnessing this response may improve anti-cancer cellular therapies.
MSCs in the tumor microenvironment
The interaction of MSCs in the tumor microenvironment is complex and driven by the interplay with numerous types including:
- Blood and lymphatic endothelial cells, which induce angiogenesis and recruitment of immune-suppressive cells, appearing as a crucial regulator of the host immune response to cancer [51,52].
- Cancer-associated fibroblasts (CAFs), which represent the predominant non-hematopoietic stromal cell type and are correlated with poor prognosis in many tumors [53,54]. CAFs have been shown to support tumor growth by providing growth factors [55] and hinder anti-tumor immune responses [56]. MSCs may contribute to this population.
- Pericytes, which differentiate from mesenchymal precursors and are recruited to tumors by platelet-derived growth factor-β (PDGF-β) [57], where they populate the luminal side of blood vessels. It is suggested that pericytes may prevent lymphocyte extravasation and activation at tumor sites [58–60].
- Tumor infiltrating leukocytes such as regulatory T cells (Treg), myeloid-derived suppressor cells (MDSC) and alternatively activated type 2 macrophages (M2). These cells create a highly immunosuppressive microenvironment with the secretion of cytokines including IL10 and TGF-β, vascular endothelial growth factor (VEGF), galectins, and expression of inhibitory receptors such as CTLA4 and PD-L1, and secretion of amino acid depleting enzymes such as arginase and IDO, PGE2. The combination of all these processes inhibits and inactivates key players of both the innate and adaptive immune system (inhibition of CD8 T cells, dendritic cells, NK cells (reviewed in [61]).
- The recruitment of MSCs to the tumor stroma has both the potential to amplify the immunosuppressive tumor microenvironment and promote tumor growth, or enhance immune properties tilting the balance towards an anti-tumor and pro-inflammatory microenvironment. Defining which molecular cues lead to each MSC-induced environment might be important to their use in clinical settings and is discussed in the following sections.
MSC immunosuppressive properties
Immune suppression by MSCs is multifactorial, occurring both by soluble signals and direct cell contact (reviewed in [62]). In vivo, MSCs produce basal levels of cytokines, adhesion molecules and inflammatory mediators, increasing their secretion of immuneosuppressive factors as well as several chemoattractants, leading to recruitment of immune cells, in response to inflammatory cytokines (IFN-γ and TNF-α). Induction of lymphocyte-specific cytokines such as CXCL9, CXCL10 and CXCL11 is dependent on the combined action of IFN-γ and TNF-α [63,64]. MSCs also express other molecules involved in feto–maternal tolerance including leukemia inhibitory factor (LIF) [65], HLA-G [66,67], and galectins 1, 3 and 8 [68,69] all of which are upregulated by exposure to IFN-γ, inhibiting T and NK cell activities. Adhesion molecules (ICAM-1 and VCAM-1) are also upregulated, though the significance of this for MSC–immune cell interactions is debated [63,70]. TSG-6 is another example of an immuneosuppressive factor upregulated in MSCs following exposure to TNF-α [71,72].
MSCs constitutively express COX-2 and low levels of PGE2, well known T-cell inhibitors, which are increased following exposure to IFN-γ or TNF-α. PGE2 is known to have an inhibitory action on T cells, and MSC-derived PGE2 has been heavily investigated and found to reduce T cell proliferation [68,73] and induce an IL10-secreting macrophage phenotype in an in vivo model of sepsis [74]. In a recent clinical trial involving lupus patients, MSC injections were suggested to inhibit Th17 polarization with the induction of a shift into IL10-producing cells and an increase in Tregs [75,76]. In another study, PGE2-secreting MSCs inhibited NK cell cytotoxicity [77]; however, other groups have shown PGE2 inhibition to have only a marginal effect on the suppression of proliferating T cells activated by MSC-secreted PGE2 [63,64,78].
Other pathways have been defined by clear cut results. Inhibition of nitric oxide (NO) (in mouse MSCs) or indoleamine 2,3-dioxygenase (IDO) (in human MSCs) totally abrogates the suppression of T-cell proliferation [63,64,79,80]. At high concentrations NO also appears to inhibit T cell activation and leads to IL10 production by macrophages. Meanwhile IDO catabolites supress NK and T cell proliferation and induce T cell apoptosis and Treg differentiation.
Other factors have also been implicated in MSC immunosuppression. MSC-derived TGF-β inhibits NK and T cells in vivo [81,82] and induces Tregs in vivo [83].
MSC immune-enhancing properties
The first of over 250 clinical trials using MSCs were performed to treat graft-versus-host disease (GvHD) patients [84]. However, therapeutic effects were not borne out and in some cases MSC administration resulted in accelerated graft rejection [85,86]. Further in vivo studies showed low dose concanavalin A or the addition of IL10, abrogated the immunosuppressive effect of MSCs [87]. Exposure of MSCs to insufficient inflammatory cytokines also results in low MSC NO secretion [88] (reviewed in [89]).
Galipeau et al showed that MSCs can act as antigen presenting cells (APCs) by engineering MSCs to stably express the kinase-inactive rat ERBB2/HER2/neu (MSC/Neu). They observed that subcutaneous injection of naïve non-activated syngeneic and allogeneic mouse MSC/Neu could induce Her-2/neu-specific T cells and antibodies, leading to the rejection of transplanted neu-expressing tumors [90].
In a different study, APC properties increased when MSCs were pre-treated with IFN-γ. However, this would only happen at low doses of IFN-γ, with higher doses pushing the MSCs towards an immunosuppressive phenotype [91]. In light of these studies, it is hypothesized that priming with IFN-γ not only increased antigen processing and presentation in MSCs but also activated them for the production of immunosuppressive factors. Importantly, these conclusions highlight the potential consequences of using MSCs as APCs in cancer immunotherapy in the presence of concomitant inflammatory response, which triggers increased antigen processing but also immune suppression by MSCs.
Inflammatory diseases play a significant role in the etiology of cancer. A variety of inflammatory cytokines and chemokines are produced within the tumor such as TNF-α, IL1, IL6 and IL8. This microenvironment will define MSC phenotype switch between immune suppressive and immune enhancing. It appears clear that MSCs switch between immunosuppressive and immune-enhancing phenotypes in response to surrounding environmental cues, and therefore a better knowledge of different tumor microenvironments and their effects on MSC phenotype will be critical for successful clinical application.
The few factors that have been elucidated thus far for the switch between immunosuppressive and immune-enhancing properties in MSCs are depicted in Figure 2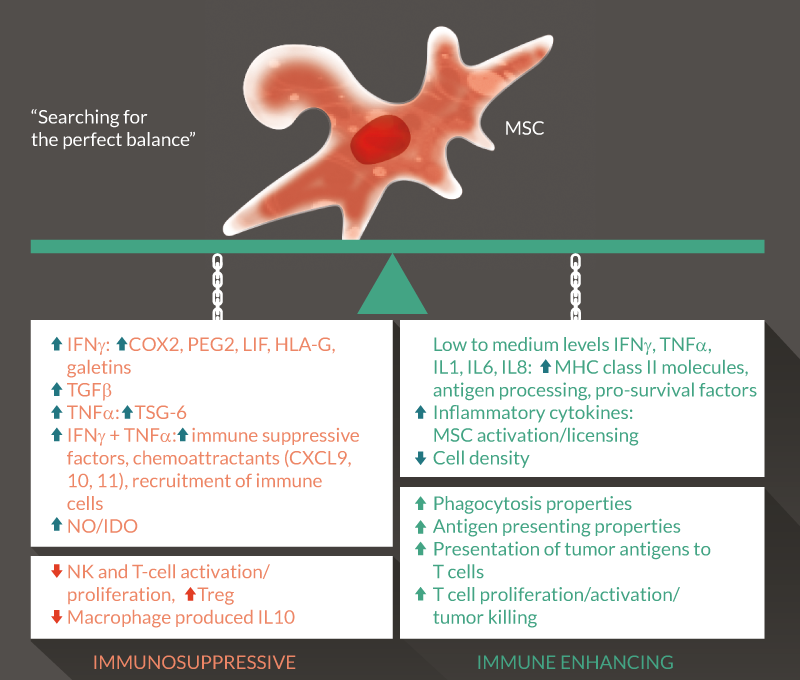
Translation Insight
Modulation of cancer by MSCs
Depending on the site and specific cancer behavior, tumors can display unique microenvironmental cues for MSCs. Both the tumor cells and the surrounding stroma release chemotactic and cytokine signals, and glucose and oxygen metabolism are often altered. MSCs respond to these cues and change their behavior accordingly.
It is highly controversial as to whether MSCs have pro- or anti-tumorigenic effects. Indeed, several conflicting in vivo and in vivo studies show MSC to either stimulate tumor cell proliferation or induce their apoptosis (reviewed in [92]).
For example, under low oxygen conditions, MSCs show increased migration ability, the capacity to form capillary-like structures [93] and secretion of high levels of VEGF in HIF-1α-dependent fashion [93]. This suggests that MSCs can be affected by the microenvironment created within hypoxic solid tumors and gain an active role in their growth.
On the one hand MSCs can potentially promote tumor growth by secreting several factors such as bFGF, VEGF, platelet-derived growth factor (PDGF), HGF, EGF receptor-ligand, insulin-like growth factor 1 (IGF-1), SDF-1 and TGF. On the other hand, MSCs have been shown to induce apoptosis in tumor cells and/or induce their growth arrest at the G1 phase of the cell cycle. This effect has been related to MSC secretion of DKK-1 [94,95], a negative regulator of Wnt/β-catenin pathway and its secretion from MSCs appears dependent on cell density [96]. In vivo models have also demonstrated discrepancies between MSC pro- and anti-tumorigenic effects. Several mouse tumor models showed an anti-tumorigenic effect from intravenously-injected MSCs (reviewed in [92]). In contrast, in other models where cells were injected subcutaneously or intraperitoneally, pro-tumorigenic effects appear more common.
Clinically only one study showed a pro-tumorigenic effect when MSCs were injected at the same time as HSC transplantation in leukemic patients [97]. It is however encouraging to note that only one of the diverse and numerous clinical trials using MSCs (45 clinical trials: https://clinicaltrials.gov; reviewed in [98]) reported tumor-promoting effects, in different settings such as HSC-transplanted patients or in patients treated for inflammatory or degenerative diseases. Over 300 clinical trials are registered using MSCs for different applications (reviewed in [99]) and 20 are registered for cancer patients (https://clinicaltrials.gov), with an emerging trend for the use of MSCs as vehicles for oncolytic viruses.
Improving MSC tumor homing for clinical applications
As discussed above, multiple different mechanisms have been implicated in MSC tumor homing, and data pertaining to these are often conflicting. Various experimental factors may account for these discrepancies, such as:
- Cell density;
- Cell expansion and the use of low versus high MSC passages;
- Differing MSC culture methods;
- Cytokines present in the model microenvironment;
- In vivo delivery route, and
- Cell dose.
Indeed, extensive passages of MSCs affected their activation and protection in an ischemia model, due to reduced secretion of growth factors [100]. Additionally, high passage MSCs tend to lose surface receptors, affecting their chemotaxis ability [101,102]. In support of this, CXCR4 has notably been shown to decrease at the cell surface after extensive passages (up to 10 in our laboratory). However, hypoxic conditions appear to enhance CXCR4 expression and increase MSC engraftment [103]. Different cocktails of cytokines can promote this effect and we have shown that tumor conditioned medium treatment can increase CXCR4 surface expression in MSC [24]. Others have also shown that pre-treatment with TNF-α, TGF-β and IL1b can stimulate MSCs to secrete high levels of matrix metalloproteinases, facilitating migration through the extracellular matrix in response to chemokines [104].
Various routes of injection have been tried, including intravenous, intraperitoneal, intra-arterial, in situ and pleural, each affecting the efficiency of homing to target organs or tumors, and therefore impacting their clinical benefit for example as anti-cancer molecule vehicles [101,105]. In general, intravenous delivery appears to be the most convenient and successful in treating certain types of diseases, although superior cell engraftment has been observed following intra-arterial and in situ injections for myocardial infarction, kidney transplantation and brain injury. We recently demonstrated in a mouse model that the delivery of the anti-cancer molecule TRAIL by MSCs reduced lung metastasis following intravenous injection, but that the same cells had no effect when administered pleurally [105].
Administration of MSCs in situ, appears less clinically attractive, as it is invasive and is the least efficient in maintaining the viability of the injected cells [106]. Other factors should not be ignored such as the stage of disease, timing of MSC delivery and number of MSCs injected, as more MSC administration does not always produce a better therapeutic effect, for example in brain injury animal models [107].
A wide range of novel techniques may be required to further elucidate MSC homing mechanisms (rolling, adhesion and transmigration), cell survival and interaction and integration within tumors and normal organs. For example, defining whether selective interactions of MSCs with tumor blood vessels or other blood vessels have a role in homing, which could be assessed with the use of intravital microscopy [108]. The use of this technique coupled to spatiotemporal FRET-based aptamer microenvironment sensors will allow the study of spatiotemporal localization of MSCs and the measure of critical signalling molecules within the tumor microenvironment [109]. The development of this wide range of novel techniques will render it possible to visualize and assess directly the interactions between MSCs and tumors in vivo.
MSCs express surface receptors capable of sensing signals released in sites of injury, inflammation or tumors. Many groups have attempted to modify MSCs to enhance surface marker expression and thereby enhance MSC migration and homing. In particular, several studies have focused on enhancing MSC expression of CXCR4. MSC transduction with CXCR4 retroviral constructs, mRNA transfection of CXCR4–GFP [110], and cytokine pre-treatment particularly with TNF-α were successful in enhancing MSC migration toward a SDF-1α gradient in vivo [111,112]. The homing receptor CCR2, highly expressed at sites of inflammation, has also been studied as a candidate for receptor enhancement. GFP-labelled CCR2-expressing MSCs were infused into transgenic mice expressing CCR2 in the myocardium. A higher number of GFP-positive cells were present in the myocardium of the transgenic mice compared to control mice [26].
Maijenburg et al used gene expression profiling to identify 12 genes differentially expressed in migratory MSCs. Within this group, the nuclear receptors Nur77 and Nurr1 were those most expressed, and pre-treatment with SDF-1α and PDGF-BB was shown to upregulate their expression. In addition, MSCs engineered to overexpress Nur77 showed increased migration in response to SDF-1α. [113].
Kumar et al transduced mouse MSCs with an adenovirus construct to upregulate the expression of the α4 subunit of VLA-4-integrin. They showed that the dimerization of this with β1-integrin improved MSC homing to bone marrow by over 10-fold in syngeneic female mice [114].
A separate, equally promising strategy for enhancing MSC homing would be to use lipid vesicles to load MSCs with surface receptors or other molecules such as SLex or P/L-selectin targeting aptamer. This circumvents difficulties seen with viral vectors in achieving effective receptor conformations and cell surface recruitment. Furthermore, the absence of genetic manipulation of the MSC product is clinically more attractive in terms of patient safety. This approach showed promising results, successfully increasing homing to inflamed endothelium, both in vivo and in vitro [109,115–117].
MSC-based therapies in clinical practice – the universal donor paradigm
It is becoming apparent that MSCs are not immune privileged, as claimed in the literature; however their use in clinical trials is escalating (https://clinicaltrials.gov/). The scientific community appears reluctant to abandon the immune privileged paradigm supporting the notion of a “universal donor” but it is clear that immunogenicity needs to be recognized as an important characteristic of MSCs.
Both syngeneic and allogeneic MSCs are currently being used in clinical trials; however very few studies focused on direct comparisons between the two. Despite the fact that both sources were deemed safe in several trials with no major adverse effects reported, two clinical trials (POSEIDON and a phase 2 mesoblast trial) observed an anti-donor response in patients treated with allogeneic MSCs [118].
The secretion of trophic and immunomodulatory factors, or the production of exosomes, immediately following MSC injection may account for their therapeutic effect by a so-called ‘hit-and-run’ mechanism [119,120]. However, this concept is challenged by the idea that the main therapeutic benefit of MSCs is achieved through reprogramming of the immune system through apoptotic bodies [121]. Nevertheless, both concepts support the consensus that a benefit of MSCs can be boosted by extending their persistence after injection [122].
A better understanding of the particular MSC mechanisms contributing to therapeutic effect in each disease setting is required in order to clarify whether allogeneic or syngeneic MSCs are more appropriate. If allogeneic MSCs were shown to possess more potent ‘hit-and-run’ effects, then one could envision their dominance in clinical settings where MSC persistence is not necessarily required. The notion that extended persistence of MSCs will result in a sustained therapeutic effect and improved clinical outcome has yet to be tested and proven clinically, depending, as discussed above, on the different disease settings and need for persistence. Nevertheless, the use of allogeneic MSC therapy and the concept of “off the shelf”, “one fits all”, needs revision and further investigation. Encouragingly, allogeneic MSC therapies have been consistently shown to be safe, allowing future trials to be conducted with improved design and standardized protocols, using refined MSC-based approaches [123]. As allogeneic MSCs appear to be cleared only marginally faster than syngeneic MSCs, combination approaches to avoid rejection and mitigate transplantation shock could be explored to extend persistence. Next-generation trials for MSC therapies should aim for in-depth characterization and fine tuning of MSC engraftment, immunogenicity, survival, potency and disease specific mechanisms of action.
Future of MSC-based therapies for cancer: where are we heading?
Oncolytic viruses, exosomes, iPS-derived MSCs
This review summarized the current challenges we face regarding the use of MSCs in a clinical setting. Our group has recently started a phase I/II clinical trial based on in vivo studies showing inhibition of tumor growth in a lung cancer model using MSCs carrying TRAIL [105].
A key area for the future will be the use of oncolytic viruses. These viruses specifically target and infect cancer cells, triggering immune activation upon cancer cell lysis. Some of the oncolytic viruses (for example vaccinia virus and vesicular stomatitis virus) are shown to inhibit the tumor immunosuppressive microenvironment, switching to an anti-tumor and pro-inflammatory microenvironment by reducing the secretion of immune-suppressive cytokines, reducing the recruitment of immune suppressive cells and inducing a vasculature shutdown leading to cancer cell necrosis (reviewed in [124,125]). A main challenge for this strategy is the clearance of the virus before it reaches the tumor bed by the host defence mechanisms. In an attempt to circumvent this issue, one strategy is to use cells as carriers, with MSCs appearing to be a good candidate.
In addition to the capacity to carry the virus to the tumors using their homing capacities, MSCs have been shown to successfully protect different oncolytic viruses from neutralizing antibodies and other host antiviral mechanisms, successfully delivering the viruses to the tumor site [126–129]. An example of one such clinical trial is the treatment of recurrent ovarian cancer using MSCs loaded with measles virus (NCT02068794).
Emerging strategies are focusing on the use of cell-free products, such as exosomes. Exosomes exert their effects via the transfer of a variety of different biomolecules (endosome-associated proteins, membrane proteins, lipid raft proteins and RNA including small interfering RNAs [siRNAs]) [130]. Exosomes isolated from MSCs have shown promising results in various animal models (reviewed in [131]). Customized production of MSC-derived exosomes is achievable. These biological nanocarriers can be loaded with drugs and siRNAs either by electroporation or chemical disruption or they can be incorporated during their biogenesis using genetically modified MSCs. The potential advantages of this strategy compared with whole MSC-based therapies include: i) higher safety profile, ii) inherent anti-inflammatory and pro-regenerative effects (which are still not fully clarified), and iii) a lower chance of rejection in allogeneic settings.
Another interesting strategy to circumvent potential safety issues following the injection of MSCs is to derive them from induced pluripotent stem cells (iPSCs). MSCs derived from iPSCs have been shown to maintain their regenerative and immunomodulatory properties [132,133]. Identification and utilization of genetically modified MSCs, having a “safe harbor” integration, is limited because of the short lifespan of primary MSCs in vivo. Using iPSCs can generate indefinite fresh MSCs. Additionally, the use of lentiviruses to modify MSCs can lead to unwanted integration and present a safety issue. Taking this issue into consideration, genetically engineered MSC clones could be generated from iPSCs after accurate screening for a vector integration site and cells with “safe harbor” integrations could potentially be expanded indefinitely, allowing the establishment of a bank of cells and an ‘off the shelf’ therapeutic product (reviewed in [134,135]).
Financial & competing interests disclosure
The authors have no relevant financial involvement with an organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock options or ownership, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.
References
1.Dominici M, Le Blanc K, Mueller I et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006; 8(4), 315–7. CrossRef
2.Samsonraj RM, Rai B, Sathiyanathan P et al. Establishing criteria for human mesenchymal stem cell potency. Stem Cells 2015; 33(6), 1878–91. CrossRef
3.Spaeth E, Klopp A, Dembinski J, Andreeff M, Marini F. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008; 15(10), 730–8. CrossRef
4.Hong HS, Lee J, Lee E et al. A new role of substance P as an injury-inducible messenger for mobilization of CD29(+) stromal-like cells. Nat. Med. 2009; 15(4), 425–35. CrossRef
5.Wang CH, Cherng WJ, Yang NI et al. Late-outgrowth endothelial cells attenuate intimal hyperplasia contributed by mesenchymal stem cells after vascular injury. Arterioscler. Thromb. Vasc. Biol. 2008; 28(1), 54–60. CrossRef
6.He Q, Wan C, Li G Concise review: multipotent mesenchymal stromal cells in blood. Stem Cells 2007; 25(1), 69–77. CrossRef
7.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007; 7(9), 678–89. CrossRef
8.Droujinine IA, Eckert MA, Zhao W. To grab the stroma by the horns: from biology to cancer therapy with mesenchymal stem cells. Oncotarget 2013; 4(5), 651–64. CrossRef
9.Teo GS, Ankrum JA, Martinelli R et al. Mesenchymal stem cells transmigrate between and directly through tumor necrosis factor-alpha-activated endothelial cells via both leukocyte-like and novel mechanisms. Stem Cells 2012; 30(11), 2472–86. CrossRef
10.Carman CV. Mechanisms for transcellular diapedesis: probing and pathfinding by ‘invadosome-like protrusions’. J. Cell Sci. 2009; 122(Pt 17), 3025–35. CrossRef
11.Ruster B, Göttig S, Ludwig RJ et al. Mesenchymal stem cells display coordinated rolling and adhesion behavior on endothelial cells. Blood 2006; 108(12), 3938–44. CrossRef
12.Segers VF, Van Riet I, Andries LJ et al. Mesenchymal stem cell adhesion to cardiac microvascular endothelium: activators and mechanisms. Am. J. Physiol. Heart Circ. Physiol. 2006; 290(4), H1370–7. CrossRef
13.Langer HF, Stellos K, Steingen C et al. Platelet derived bFGF mediates vascular integrative mechanisms of mesenchymal stem cells in vitro. J. Mol. Cell Cardiol. 2009; 47(2), 315–25. CrossRef
14.Schmidt A, Ladage D, Steingen C et al. Mesenchymal stem cells transmigrate over the endothelial barrier. Eur J Cell Biol 2006; 85(11), 1179–88. CrossRef
15.Steingen C, Brenig F, Baumgartner L, Schmidt J, Schmidt A, Bloch W. Characterization of key mechanisms in transmigration and invasion of mesenchymal stem cells. J. Mol. Cell Cardiol. 2008; 44(6), 1072–84. CrossRef
16.Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat. Immunol. 2005; 6(12), 1182–90. CrossRef
17.Ara T, Tokoyoda K, Sugiyama T, Egawa T, Kawabata K, Nagasawa T. Long-term hematopoietic stem cells require stromal cell-derived factor-1 for colonizing bone marrow during ontogeny. Immunity 2003; 19(2), 257–67. CrossRef
18.Geminder H, Sagi-Assif O, Goldberg L et al. A possible role for CXCR4 and its ligand, the CXC chemokine stromal cell-derived factor-1, in the development of bone marrow metastases in neuroblastoma. J. Immunol. 2001; 167(8), 4747–57. CrossRef
19.Muller A, Homey B, Soto H et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001; 410(6824), 50–6. CrossRef
20.Porcile C, Bajetto A, Barbero S, Pirani P, Schettini G. CXCR4 activation induces epidermal growth factor receptor transactivation in an ovarian cancer cell line. Ann. NY Acad. Sci. 2004; 1030: 162–9. CrossRef
21.Sun YX, Wang J, Shelburne CE et al. Expression of CXCR4 and CXCL12 (SDF-1) in human prostate cancers (PCa) in vivo. J. Cell Biochem. 2003; 89(3), 462–73. CrossRef
22.Gao H, Priebe W, Glod J, Banerjee D. Activation of signal transducers and activators of transcription 3 and focal adhesion kinase by stromal cell-derived factor 1 is required for migration of human mesenchymal stem cells in response to tumor cell-conditioned medium. Stem Cells 2009; 27(4), 857–65. CrossRef
23.Phillips RJ, Burdick MD, Lutz M, Belperio JA, Keane MP, Strieter RM. The stromal derived factor-1/CXCL12-CXC chemokine receptor 4 biological axis in non-small cell lung cancer metastases. Am. J. Respir. Crit. Care Med. 2003; 167(12), 1676–86. CrossRef
24.Lourenco S, Teixeira VH, Kalber T, Jose RJ, Floto RA, Janes SM. Macrophage migration inhibitory factor-CXCR4 is the dominant chemotactic axis in human mesenchymal stem cell recruitment to tumors. J. Immunol. 2015; 194(7), 3463–74. CrossRef
25.Komarova S, Roth J, Alvarez R, Curiel DT, Pereboeva L. Targeting of mesenchymal stem cells to ovarian tumors via an artificial receptor. J. Ovarian Res. 2010; 3: 12. CrossRef
26.Belema-Bedada F, Uchida S, Martire A, Kostin S, Braun T. Efficient homing of multipotent adult mesenchymal stem cells depends on FROUNT-mediated clustering of CCR2. Cell Stem Cell 2008; 2(6), 566–75. CrossRef
27.Dwyer RM, Potter-Beirne SM, Harrington KA et al. Monocyte chemotactic protein-1 secreted by primary breast tumors stimulates migration of mesenchymal stem cells. Clin. Cancer Res. 2007; 13(17), 5020–7. CrossRef
28.Kim SM, Kim DS, Jeong CH et al. CXC chemokine receptor 1 enhances the ability of human umbilical cord blood-derived mesenchymal stem cells to migrate toward gliomas. Biochem. Biophys. Res. Commun. 2011; 407(4), 741–6. CrossRef
29.Rattigan Y, Hsu JM, Mishra PJ, Glod J, Banerjee D. Interleukin 6 mediated recruitment of mesenchymal stem cells to the hypoxic tumor milieu. Exp. Cell Res. 2010; 316(20), 3417–24. CrossRef
30.Binsky I, Haran M, Starlets D et al. IL-8 secreted in a macrophage migration-inhibitory factor- and CD74-dependent manner regulates B cell chronic lymphocytic leukemia survival. Proc. Natl Acad. Sci. USA 2007; 104(33), 13408–13. CrossRef
31.Cheng Q, McKeown SJ, Santos L, Santiago FS, Khachigian LM, Morand EF, Hickey MJ. Macrophage migration inhibitory factor increases leukocyte-endothelial interactions in human endothelial cells via promotion of expression of adhesion molecules. J. Immunol. 2010; 185(2), 1238–47. CrossRef
32.Kim HR, Park MK, Cho ML et al. Macrophage migration inhibitory factor upregulates angiogenic factors and correlates with clinical measures in rheumatoid arthritis. J. Rheumatol. 2007; 34(5), 927–36.
33.Onodera S, Nishihira J, Koyama Y, et al. Macrophage migration inhibitory factor up-regulates the expression of interleukin-8 messenger RNA in synovial fibroblasts of rheumatoid arthritis patients: common transcriptional regulatory mechanism between interleukin-8 and interleukin-1beta. Arthritis Rheum. 2004; 50(5), 1437–47. CrossRef
34.Barbero S, Bonavia R, Bajetto A et al. Stromal cell-derived factor 1a stimulates human glioblastoma cell growth through the activation of both extracellular signal-regulated kinases 1/2 and Akt. Cancer Res. 2003; 63(8), 1969–74. CrossRef
35.Ehtesham M, Yuan X, Kabos P et al. Glioma tropic neural stem cells consist of astrocytic precursors and their migratory capacity is mediated by CXCR4. Neoplasia 2004; 6(3), 287–93. CrossRef
36.Bando H, Matsumoto G, Bando M, et al. Expression of macrophage migration inhibitory factor in human breast cancer: association with nodal spread. Jpn J. Cancer Res. 2002; 93(4), 389–96. CrossRef
37.Kamimura A, Kamachi M, Nishihira J et al. Intracellular distribution of macrophage migration inhibitory factor predicts the prognosis of patients with adenocarcinoma of the lung. Cancer 2000; 89(2), 334–41. CrossRef
38.Markert JM, Fuller CM, Gillespie GY et al. Differential gene expression profiling in human brain tumors. Physiol. Genomics 2001; 5(1), 21–33. CrossRef
39.Meyer-Siegler K, Fattor RA, Hudson PB. Expression of macrophage migration inhibitory factor in the human prostate. Diagn. Mol. Pathol. 1998; 7(1), 44–50. CrossRef
40.Shimizu T, Abe R, Nakamura H, Ohkawara A, Suzuki M, Nishihira J. High expression of macrophage migration inhibitory factor in human melanoma cells and its role in tumor cell growth and angiogenesis. Biochem. Biophys. Res. Commun. 1999; 264(3), 751–8. CrossRef
41.Takahashi N, Nishihira J, Sato Y et al. Involvement of macrophage migration inhibitory factor (MIF) in the mechanism of tumor cell growth. Mol. Med. 1998; 4(11), 707–14. CrossRef
42.Winner M, Leng L, Zundel W, Mitchell RA. Macrophage migration inhibitory factor manipulation and evaluation in tumoral hypoxic adaptation. Methods Enzymol. 2007; 435: 355–69. CrossRef
43.Baron N, Deuster O, Noelker C et al. Role of macrophage migration inhibitory factor in primary glioblastoma multiforme cells. J. Neurosci. Res. 2011; 89(5), 711–7. CrossRef
44.Ankrum JA, Ong J, Karp JM. Mesenchymal stem cells: immune evasive, not immune privileged. Nat. Biotechnol. 2014; 32(3), 252–60. CrossRef
45.Zangi L, Margalit R, Reich-Zeliger S et al. Direct imaging of immune rejection and memory induction by allogeneic mesenchymal stromal cells. Stem Cells 2009; 27(11), 2865–74. CrossRef
46.Badillo AT, Beggs KJ, Javazon EH, Tebbets JC, Flake AW. Murine bone marrow stromal progenitor cells elicit an in vivo cellular and humoral alloimmune response. Biol. Blood Marrow Transplant. 2007; 13(4), 412–22. CrossRef
47.Nauta AJ, Westerhuis G, Kruisselbrink AB, Lurvink EG, Willemze R, Fibbe WE. Donor-derived mesenchymal stem cells are immunogenic in an allogeneic host and stimulate donor graft rejection in a nonmyeloablative setting. Blood 2006; 108(6), 2114–20. CrossRef
48.Le Blanc K, Tammik C, Rosendahl K, Zetterberg E, Ringdén O. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp. Hematol 2003; 31(10), 890–6. CrossRef
49.Francois M, Romieu-Mourez R, Stock-Martineau S, Boivin MN, Bramson JL, Galipeau J. Mesenchymal stromal cells cross-present soluble exogenous antigens as part of their antigen-presenting cell properties. Blood 2009; 114(13), 2632–8. CrossRef
50.Park D, Spencer JA, Koh BI, et al. Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell 2012; 10(3), 259–72. CrossRef
51.Adotevi O, Pere H, Ravel P et al. A decrease of regulatory T cells correlates with overall survival after sunitinib-based antiangiogenic therapy in metastatic renal cancer patients. J. Immunother. 2010; 33(9), 991–8. CrossRef
52.Ko JS, Zea AH, Rini BI et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin. Cancer Res. 2009; 15(6), 2148–57. CrossRef
53.Henry LR, Lee HO, Lee JS et al. Clinical implications of fibroblast activation protein in patients with colon cancer. Clin. Cancer Res. 2007; 13(6), 1736–41. CrossRef
54.Tsujino T, Seshimo I, Yamamoto H et al. Stromal myofibroblasts predict disease recurrence for colorectal cancer. Clin. Cancer Res. 2007; 13(7), 2082–90. CrossRef
55.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature 2004; 432(7015), 332–7. CrossRef
56.Straussman R, Morikawa T, Shee K et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012; 487(7408), 500–4. CrossRef
57.Abramsson A1, Lindblom P, Betsholtz C. Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J. Clin. Invest. 2003; 112(8), 1142–51. CrossRef
58.Hamzah J, Jugold M, Kiessling F et al. Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature 2008; 453(7193), 410–4. CrossRef
59.Bose A, Barik S, Banerjee S et al. Tumor-derived vascular pericytes anergize Th cells. J. Immunol. 2013; 191(2), 971–81. CrossRef
60.Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 2007; 179(6), 1311–23. CrossRef
61.Devaud C, John LB, Westwood JA, Darcy PK, Kershaw MH. Immune modulation of the tumor microenvironment for enhancing cancer immunotherapy. Oncoimmunology 2013; 2(8), e25961. CrossRef
62.Stagg J & Galipeau J. Mechanisms of immune modulation by mesenchymal stromal cells and clinical translation. Curr. Mol. Med. 2013; 13(5), 856–67. CrossRef
63.Ren G, Su J, Zhang L et al. Species variation in the mechanisms of mesenchymal stem cell-mediated immunosuppression. Stem Cells 2009; 27(8), 1954–62. CrossRef
64.Ren G, Zhang L, Zhao X et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell 2008; 2(2), 141–50. CrossRef
65.Nasef A, Mazurier C, Bouchet S et al. Leukemia inhibitory factor: Role in human mesenchymal stem cells mediated immunosuppression. Cell Immunol. 2008; 253(1–2), 16–22. CrossRef
66.Nasef A, Mathieu N, Chapel A et al. Immunosuppressive effects of mesenchymal stem cells: involvement of HLA-G. Transplantation 2007; 84(2), 231–7. CrossRef
67.Selmani Z, Naji A, Zidi I et al. Human leukocyte antigen-G5 secretion by human mesenchymal stem cells is required to suppress T lymphocyte and natural killer function and to induce CD4+CD25highFOXP3+ regulatory T cells. Stem Cells 2008; 26(1), 212–22. CrossRef
68.Najar M, Raicevic G, Id Boufker H, et al. Modulated expression of adhesion molecules and galectin-1: role during mesenchymal stromal cell immunoregulatory functions. Exp. Hematol. 2010; 38(10), 922–32. CrossRef
69.Sioud M, Mobergslien A, Boudabous A, Fløisand Y. Evidence for the involvement of galectin-3 in mesenchymal stem cell suppression of allogeneic T-cell proliferation. Scand J. Immunol. 2010; 71(4), 267–74. CrossRef
70.Ren G, Zhao X, Zhang L et al. Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression. J. Immunol. 2010; 184(5), 2321–8. CrossRef
71.Lee RH, Pulin AA, Seo MJ et al. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell 2009; 5(1), 54–63. CrossRef
72.Roddy GW, Oh JY, Lee RH et al. Action at a distance: systemically administered adult stem/progenitor cells (MSCs) reduce inflammatory damage to the cornea without engraftment and primarily by secretion of TNF-alpha stimulated gene/protein 6. Stem Cells 2011; 29(10), 1572–9. CrossRef
73.Aggarwal S & Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005; 105(4), 1815–22. CrossRef
74.Nemeth K, Leelahavanichkul A, Yuen PS et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat. Med. 2009; 15(1), 42–9. CrossRef
75.Ghannam S, Pène J, Moquet-Torcy G, Jorgensen C, Yssel H. Mesenchymal stem cells inhibit human Th17 cell differentiation and function and induce a T regulatory cell phenotype. J. Immunol. 2010; 185(1), 302–12. CrossRef
76.Sun L, Wang D, Liang J et al. Umbilical cord mesenchymal stem cell transplantation in severe and refractory systemic lupus erythematosus. Arthritis Rheum. 2010; 62(8), 2467–75. CrossRef
77.Spaggiari GM, Capobianco A, Abdelrazik H, Becchetti F, Mingari MC, Moretta L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood 2008; 111(3), 1327–33. CrossRef
78.Rasmusson I, Ringdén O, Sundberg B, Le Blanc K. Mesenchymal stem cells inhibit lymphocyte proliferation by mitogens and alloantigens by different mechanisms. Exp. Cell Res. 2005; 305(1), 33–41. CrossRef
79.Meisel R, Zibert A, Laryea M, Göbel U, Däubener W, Dilloo D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood 2004; 103(12), 4619–21. CrossRef
80.Sato K, Ozaki K, Oh I et al. Nitric oxide plays a critical role in suppression of T-cell proliferation by mesenchymal stem cells. Blood 2007; 109(1), 228–34. CrossRef
81.Di Nicola M, Carlo-Stella C, Magni M et al. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 2002; 99(10), 3838–43. CrossRef
82.Sotiropoulou PA, Perez SA, Gritzapis AD, Baxevanis CN, Papamichail M. Interactions between human mesenchymal stem cells and natural killer cells. Stem Cells 2006; 24(1), 74–85. CrossRef
83.Patel SA, Meyer JR, Greco SJ, Corcoran KE, Bryan M, Rameshwar P. Mesenchymal stem cells protect breast cancer cells through regulatory T cells: role of mesenchymal stem cell-derived TGF-beta. J. Immunol. 2010; 184(10), 5885–94. CrossRef
84.Le Blanc K, Rasmusson I, Sundberg B et al. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet 2004; 363(9419), 1439–41. CrossRef
85.Inoue S, Popp FC, Koehl GE et al. Immunomodulatory effects of mesenchymal stem cells in a rat organ transplant model. Transplantation 2006; 81(11), 1589–95. CrossRef
86.Kuo YR, Goto S, Shih HS et al. Mesenchymal stem cells prolong composite tissue allotransplant survival in a swine model. Transplantation 2009; 87(12), 1769–77. CrossRef
87.Renner P, Eggenhofer E, Rosenauer A et al. Mesenchymal stem cells require a sufficient, ongoing immune response to exert their immunosuppressive function. Transplant Proc. 2009; 41(6), 2607–11. CrossRef
88.Li W, Ren G, Huang Y et al. Mesenchymal stem cells: a double-edged sword in regulating immune responses. Cell Death Differ. 2012; 19(9), 1505–13. CrossRef
89.Ma S, Xie N, Li W, Yuan B, Shi Y, Wang Y. Immunobiology of mesenchymal stem cells. Cell Death Differ. 2014; 21(2), 216–25. CrossRef
90.Romieu-Mourez R, François M, Abate A et al. Mesenchymal stromal cells expressing ErbB-2/neu elicit protective antibreast tumor Immunity in vivo, which is paradoxically suppressed by IFN-gamma and tumor necrosis factor-alpha priming. Cancer Res. 2010; 70(20), 7742–7. CrossRef
91.Chan JL, Tang KC, Patel AP, Bonilla LM, Pierobon N, Ponzio NM, Rameshwar P. Antigen-presenting property of mesenchymal stem cells occurs during a narrow window at low levels of interferon-gamma. Blood 2006; 107(12), 4817–24. CrossRef
92.Klopp AH, Gupta A, Spaeth E, Andreeff M, Marini F. Concise review: Dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth? Stem Cells 2011; 29(1), 11–9. CrossRef
93.Annabi B, Lee YT, Turcotte S et al. Hypoxia promotes murine bone-marrow-derived stromal cell migration and tube formation. Stem Cells 2003; 21(3), 337–47. CrossRef
94.Qiao L, Xu ZL, Zhao TJ, Ye LH, Zhang XD. Dkk-1 secreted by mesenchymal stem cells inhibits growth of breast cancer cells via depression of Wnt signalling. Cancer Lett. 2008; 269(1), 67–77. CrossRef
95.Zhu Y, Sun Z, Han Q et al. Human mesenchymal stem cells inhibit cancer cell proliferation by secreting DKK-1. Leukemia 2009; 23(5), 925–33. CrossRef
96.Gregory CA, Singh H, Perry AS, Prockop DJ. The Wnt signaling inhibitor dickkopf-1 is required for reentry into the cell cycle of human adult stem cells from bone marrow. J. Biol. Chem. 2003; 278(30), 28067–78. CrossRef
97.Ning H, Yang F, Jiang M et al. The correlation between cotransplantation of mesenchymal stem cells and higher recurrence rate in hematologic malignancy patients: outcome of a pilot clinical study. Leukemia 2008; 22(3), 593–9. CrossRef
98.Kim N & Cho SG. Clinical applications of mesenchymal stem cells. Korean J. Intern. Med. 2013; 28(4), 387–402. CrossRef
99.Wei X, Yang X, Han ZP, Qu FF, Shao L, Shi YF. Mesenchymal stem cells: a new trend for cell therapy. Acta Pharmacol. Sin. 2013; 34(6), 747–54. CrossRef
100.Crisostomo PR, Wang M, Wairiuko GM et al. High passage number of stem cells adversely affects stem cell activation and myocardial protection. Shock 2006; 26(6), 575–80. CrossRef
101.Karp JM & Leng Teo GS. Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell 2009; 4(3), 206–16. CrossRef
102.Rombouts WJ & Ploemacher RE. Primary murine MSC show highly efficient homing to the bone marrow but lose homing ability following culture. Leukemia 2003; 17(1), 160–70. CrossRef
103.Hung SC, Pochampally RR, Hsu SC et al. Short-term exposure of multipotent stromal cells to low oxygen increases their expression of CX3CR1 and CXCR4 and their engraftment in vivo. PLoS One 2007; 2(5), e416. CrossRef
104.Sordi V, Malosio ML, Marchesi F et al. Bone marrow mesenchymal stem cells express a restricted set of functionally active chemokine receptors capable of promoting migration to pancreatic islets. Blood 2005; 106(2), 419–27. CrossRef
105.Sage EK, Kolluri KK, McNulty K et al. Systemic but not topical TRAIL-expressing mesenchymal stem cells reduce tumour growth in malignant mesothelioma. Thorax 2014; 69(7), 638–47. CrossRef
106.Muschler GF, Nakamoto C, Griffith LG. Engineering principles of clinical cell-based tissue engineering. J. Bone Joint Surg. Am. 2004; 86-A(7), 1541–58. CrossRef
107.Wu J, Sun Z, Sun HS et al. Intravenously administered bone marrow cells migrate to damaged brain tissue and improve neural function in ischemic rats. Cell Transplant 2008; 16(10), 993–1005. CrossRef
108.Jain RK, Munn LL, Fukumura D. Dissecting tumour pathophysiology using intravital microscopy. Nat. Rev. Cancer 2002; 2(4), 266–76. CrossRef
109.Zhao W, Schafer S, Choi J et al. Cell-surface sensors for real-time probing of cellular environments. Nat. Nanotechnol. 2011; 6(8), 524–31. CrossRef
110.Ryser MF, Ugarte F, Thieme S, Bornhäuser M, Roesen-Wolff A, Brenner S. mRNA transfection of CXCR4-GFP fusion–simply generated by PCR-results in efficient migration of primary human mesenchymal stem cells. Tissue Eng. Part C Methods 2008; 14(3), 179–84. CrossRef
111.Baek SJ, Kang SK, Ra JC. In vitro migration capacity of human adipose tissue-derived mesenchymal stem cells reflects their expression of receptors for chemokines and growth factors. Exp. Mol. Med. 2011; 43(10), 596–603. CrossRef
112.Xiao Q, Wang SK, Tian H et al. TNF-alpha increases bone marrow mesenchymal stem cell migration to ischemic tissues. Cell Biochem. Biophys. 2012; 62(3), 409–14. CrossRef
113.Maijenburg MW, Gilissen C, Melief SM et al. Nuclear receptors Nur77 and Nurr1 modulate mesenchymal stromal cell migration. Stem Cells Dev. 2012; 21(2), 228–38. CrossRef
114.Kumar S & Ponnazhagan S. Bone homing of mesenchymal stem cells by ectopic alpha 4 integrin expression. FASEB J. 2007; 21(14), 3917–27. CrossRef
http://dx.doi.org/10.1096/fj.07-8275com
115.Sarkar D, Spencer JA, Phillips JA et al. Engineered cell homing. Blood 2011; 118(25), e184–91. CrossRef
116.Sarkar D, Vemula PK, Teo GS, et al. Chemical engineering of mesenchymal stem cells to induce a cell rolling response. Bioconjug. Chem. 2008; 19(11), 2105–9. CrossRef
117.Sarkar D, Vemula PK, Zhao W, Gupta A, Karnik R, Karp JM. Engineered mesenchymal stem cells with self-assembled vesicles for systemic cell targeting. Biomaterials 2010; 31(19), 5266–74. CrossRef
118.Hare JM, Fishman JE, Gerstenblith G et al. Comparison of allogeneic vs autologous bone marrow-derived mesenchymal stem cells delivered by transendocardial injection in patients with ischemic cardiomyopathy: the POSEIDON randomized trial. JAMA 2012; 308(22), 2369–79. CrossRef
119.Prockop DJ. Concise review: two negative feedback loops place mesenchymal stem/stromal cells at the center of early regulators of inflammation. Stem Cells 2013; 31(10), 2042–6. CrossRef
120.von Bahr L, Batsis I, Moll G et al. Analysis of tissues following mesenchymal stromal cell therapy in humans indicates limited long-term engraftment and no ectopic tissue formation. Stem Cells 2012; 30(7), 1575–8. CrossRef
121.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature 1997; 390(6658), 350–1. CrossRef
122.Ranganath SH, Levy O, Inamdar MS, Karp JM. Harnessing the mesenchymal stem cell secretome for the treatment of cardiovascular disease. Cell Stem Cell 2012; 10(3), 244–58. CrossRef
123.Ankrum J & Karp JM. Mesenchymal stem cell therapy: Two steps forward, one step back. Trends Mol. Med. 2010; 16(5), 203–9. CrossRef
124.Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015; 14(9), 642–62. CrossRef
125.Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014; 14(8), 559–67. CrossRef
126.Castleton A, Dey A, Beaton B et al. Human mesenchymal stromal cells deliver systemic oncolytic measles virus to treat acute lymphoblastic leukemia in the presence of humoral immunity. Blood 2014; 123(9), 1327–35. CrossRef
127.Hoyos V, Del Bufalo F, Yagyu S et al, Mesenchymal Stromal Cells for Linked Delivery of Oncolytic and Apoptotic Adenoviruses to Non-small-cell Lung Cancers. Mol. Ther. 2015; 23(9), 1497–506. CrossRef
128.Leoni V, Gatta V, Palladini A et al. Systemic delivery of HER2-retargeted oncolytic-HSV by mesenchymal stromal cells protects from lung and brain metastases. Oncotarget 2015; 6(33), 34774–87.
129.Duebgen M, Martinez-Quintanilla J, Tamura K et al. Stem cells loaded with multimechanistic oncolytic herpes simplex virus variants for brain tumor therapy. J. Natl Cancer Inst. 2014; 106(6), dju090. CrossRef
130.Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007; 9(6), 654–9. CrossRef
131.Rani S, Ryan AE, Griffin MD, Ritter T. Mesenchymal Stem Cell-derived Extracellular Vesicles: Toward Cell-free Therapeutic Applications. Mol. Ther. 2015; 23(5), 812–23. CrossRef
132.Moslem M, Valojerdi MR, Pournasr B, Muhammadnejad A, Baharvand H. Therapeutic potential of human induced pluripotent stem cell-derived mesenchymal stem cells in mice with lethal fulminant hepatic failure. Cell Transplant. 2013; 22(10), 1785–99. CrossRef
133.Sun YQ, Deng MX, He J et al. Human pluripotent stem cell-derived mesenchymal stem cells prevent allergic airway inflammation in mice. Stem Cells 2012; 30(12), 2692–9. CrossRef
134.Jung Y, Bauer G, Nolta JA. Concise review: Induced pluripotent stem cell-derived mesenchymal stem cells: progress toward safe clinical products. Stem Cells 2012; 30(1), 42–7. CrossRef
135.Focosi D, Amabile G, Di Ruscio A, Quaranta P, Tenen DG, Pistello M. Induced pluripotent stem cells in hematology: current and future applications. Blood Cancer J. 2014; 4: e211. CrossRef
Affiliations
Sofia Lourenco, Elizabeth F Maughan & Sam M Janes
Lungs for Living Research Centre,
Division of Medicine,
University College London,
London,
WC1E 6JF, UK