The promise of therapeutic genome engineering
Cell Gene Therapy Insights 2015; 1(2), 195-214
10.18609.cgti.2015.017
Recent advances in genome engineering have revolutionized our ability to specifically and delicately manipulate the genomes of essentially any organism including human cells, and have already led to more accurate cellular and animal models of disease. Such techniques also have the potential to permanently repair genetic mutations associated with human disease, many of which are currently difficult or impossible to treat by traditional means. This review discusses the technologies currently available for genome engineering, the strategies for their application in patients and current progress towards applying such techniques to specific diseases. I highlight the exciting avenues for such therapeutic genome engineering in the future and challenges to its successful application.
Sequencing of the human genome has provided us with the means to perform detailed analysis of the genetic variants underlying many diseases, and to date nearly 3,500 genes have been linked to specific pathologies (www.omim.org/statistics/geneMap). Certain genetic variants have been shown to be causative in disease through familial inheritance studies. Such well-defined monogenic disorders, including cystic fibrosis, hemophilia B, severe combined immunodeficiency (SCID), and sickle cell anemia provide good candidates for therapeutic genetic intervention. Since they are largely caused by mutation within a single gene, correction of this aberration would therefore be expected to completely revert the pathological symptoms.
Whilst the underlying genetics of many diseases have been well studied, our ability to intervene in this process has, until recently, been limited. Techniques such as developing small molecules to predicted target genes, oligonucleotide-based approaches to interfere with gene function, expression of transgenes to restore dysfunctional gene expression or knockdown of gene expression post-transcriptionally by RNA interference (RNAi) have been applied (Reviewed in [1,2]). However, these techniques have certain limitations, since they need to be constantly supplied to the tissue of interest, and often require integration of foreign DNA into the genome, which can result in undesirable mutations [3]. The recent explosion in genome engineering techniques such as zinc finger nucleases (ZFNs [4]), transcription activator-like effector nucleases (TALENs [5]) and clustered regularly interspaced short palindromic repeat associated endonucleases (CRISPR/Cas9 [6]) have allowed delicate and specific manipulation of the DNA sequence. This has not only enabled us to more rapidly generate cellular and animal models of genetic variants, but also opened up the possibility of genetic therapies to permanently correct disease-causing aberrations both somatically and more controversially in the germline [7,8].
As well as treatment for heritable genetic diseases, genome engineering may also provide an attractive alternative to treat infectious diseases. The exquisite sequence-specific binding of these reagents can be used to target viral and bacterial genomes, to prevent the spread of specific subtypes of pathogen such as hepatitis B virus [9–14] and bacteria such as antibiotic-resistant Staphylococcus aureus [15,16]. Interestingly, such sequence-specific reagents can also be applied to cure latent viral infections, where the virus has integrated into the genome of the host cell, such as in the case of HIV [17–19].
These recent developments in the identification of genetic variants associated with disease and the genome engineering technologies necessary for their correction may therefore realize our ambition of being able to correct genetic aberrations that cause a variety of debilitating diseases.
Overview of systems
Zinc fingers
The concept of genome engineering, that is the use of specific DNA-binding factors to manipulate DNA sequence, was born over 20 years ago with the discovery that ZFN DNA-binding transcription factors were modular in their architecture [20,21]. This led to the proposal that they could be reprogrammed to bind to essentially any sequence, and carry with them DNA-modifying enzymes to introduce mutations or other modifications to the underlying DNA [20,21]. Each ZFN monomer was shown to bind to three consecutive bases in the DNA, but frustratingly the DNA sequence bound by a single monomer differs depending on its context within the polypeptide [22,23]. This makes prediction of their binding sites challenging, and it is therefore necessary to invoke elaborate selection strategies from libraries of ZFN multimers to identify those that bind to the desired sequence, hindering general application of this technique [24]. Certain sequences, notably those deficient in guanine bases, are also inherently more difficult to target simply due to the absence of monomers that are able to bind to these sequences.
These DNA-binding domains are often fused to a non-specific endonuclease domain (e.g., Fok I [25]), and used to introduce mutagenic double strand breaks (DSBs) in the genome (Figure 1). In order to increase specificity and avoid unwanted DSBs, typically two ZFN DNA-binding domains are produced, each of which carries half of a homo- or hetero-dimeric Fok I nuclease [26,27]. The nuclease therefore only becomes functional upon binding of two ZFNs in the correct orientation and spacing on the DNA, improving specificity considerably, but also further limiting the potential target sites within the genome.

Schematics of different DNA- binding factors are indicated, along with information about DNA recognition site, sequence constraints on-target site choice, off-targeting, ease of assembly, in vivo delivery and multiplexing. AAV: adeno-associated virus; PAM: protospacer adjacent motif.
TALEs
A second class of highly modular DNA-binding factors, the transcription activator like effector (TALE) proteins, were discovered through studies of the plant pathogen Xanthomonas, which uses them to modulate host gene transcription [28–30]. TALEs are made up of repeating 34-amino-acid modules, each of which binds to a single base in the DNA, the identity of which is determined by two amino acids, the repeat variable diresidue (RVD), in each monomer [28,30]. Since each monomer acts independently of its location within the polypeptide, it is possible to predict the sequence of monomers necessary to bind to essentially any DNA sequence [31,32]. In order to achieve specificity within a large genome, polypeptides with up to 20 monomers are necessary, which along with their inherently repetitive nature, makes assembly and manipulation of these proteins somewhat challenging. Although elegant systems have been developed that allow the production of such polypeptides [31,33,34], this process is still somewhat laborious, making assembly of large numbers of constructs time consuming.
Similar to ZFNs, these DNA-binding domains are often designed in pairs, and fused to homo- or hetero-dimeric endonuclease domains to generate TALE nucleases or TALENs [31,32,35]. Unlike ZFNs, they can be targeted to any DNA sequence so long as the first base bound by each TALE is a thymine, and lengths of multimers can be adjusted to provide sufficient specificity (Figure 1).
CRISPR/Cas9
Recent studies on bacterial immunity to viral infection led to the exciting discovery of a class of RNA-guided endonucleases present in many different bacterial species [36–39]. Endogenously, viral fragments are captured into arrays of CRISPR, that are subsequently transcribed into CRISPR RNAs (crRNAs) which bind to the CRISPR-associated endonuclease (Cas9) protein and guide it to specific sites in the viral genome, where it generates mutagenic DSBs. The realization that the sequence specificity was driven by Watson–Crick base pairing of the first 20 nt of the guide RNA molecule with the DNA target site made it possible to reprogram the endonuclease to essentially any target site in a highly predictable manner simply by altering this 20 nt sequence [40–42](Figure 1). The most commonly used system derives from Streptococcus pyogenes, and can be recapitulated by simply expressing two components: an ~100 nt synthetic guide RNA (sgRNA) and the Cas9 protein. This has led to the demonstration of its activity in many other organisms, including human cells [43–45] and cynomolgus monkeys [46], and its rapid exploitation in model and non-model organisms [47–49]. The simplicity of the system allows the extremely rapid assembly of large numbers of constructs, and combining this with techniques used to generate oligonucleotide arrays has allowed genome-wide libraries of up to 200,000 guides to be produced, targeting every annotated gene in the human genome [50,51].
The only limitation to the sequences that can be targeted is the necessity for a protospacer adjacent motif (PAM) adjacent to the guide RNA, that is recognized by the Cas9 protein [52]. For the S. pyogenes system this is NGG, which should occur on average every eight bases in the DNA. However, CRISPR systems from other bacteria have different PAM requirements [53–55], and there has been success in engineering Cas proteins to utilize different PAM sequences [56], making it likely that in the future it will be possible to target essentially any sequence.
Specificity of Cas9 binding is potentially a problem, especially in large genomes such as in humans, since it is determined by a 20 nt sequence and the requirement for the PAM (NGG). As with all of the genome engineering systems described, there is some tolerance for mismatches, making the situation worse. However, in the case of CRISPR/Cas9, this is somewhat more problematic, since the effect of mismatches is position dependent, and 2–3 mismatches at the 5′ end of the sgRNA can be tolerated [57,58] (Figure 1). Whilst the exact nature and extent of off-targeting is currently under debate [57,59–65], and likely depends on cell type and delivery system, there are several systems that have been developed to address this problem. A single point mutation in Cas9 (D10A) results in inactivation of one of the two endonuclease domains, preventing cleavage of one DNA strand and turning the enzyme into a “nickase”, only able to make single strand DNA (ssDNA) breaks, which are rapidly and efficiently repaired by the cell [58,66]. However, supplying two guides in an appropriate orientation and spacing allows a staggered DSB to be made, that results in similar mutagenic rates to the wild-type protein. Similarly, a catalytically dead Cas9 protein (D10AH840A) fused to the dimeric Fok I nuclease allows one to employ an analogous system to that used for the ZFNs and TALENs, with a pair of guide RNAs driving specificity [67]. Somewhat unexpectedly, specificity can also be improved by reducing the length of the guide RNA without significantly reducing catalytic activity at least in some cases [68,69]. Whilst these techniques already vastly improve specificity, no doubt additional improvements will continue to develop through engineering of sgRNA sequence, Cas9 protein, chemical modifications and delivery systems. The recent description of Cas9 structures in complex with sgRNA and target DNA will no doubt facilitate such efforts [70,71].
Approaches to use
Predominantly, genome engineering systems are used to introduce genetic mutations at desired sites in the genome. This has already enabled more rapid genetic analysis in model organisms [47–49] and allowed genetic modifications to be performed in systems that would otherwise not be amenable to such manipulations [43–46,48].
Such genetic changes can be highly beneficial for therapeutic applications (Table 1), since it is possible to effect a permanent repair to the DNA, that persists for the lifetime of the cell, and does not require continued transgene expression after the repair has been completed. This is achieved by targeting DSBs to desired sites in the DNA, and exploiting the endogenous DNA repair mechanisms of non-homologous end joining (NHEJ) or homologous recombination (HR), to generate the desired changes in the DNA [72,73].
| Hereditary disease | Target gene | Nuclease | Strategy | in vivo/ex vivo | Refs |
|---|---|---|---|---|---|
| Cystic fibrosis | CFTR | CRISPR | HR-based repair of CFTR gene in intestinal organoids | ex vivo | [113] |
| Hemophilia B | Factor IX | ZnF | HR-based repair of Factor IX gene in liver cells | in vivo | [97] |
| SCID | IL2RG | ZnF | HR-based repair of IL2RG gene in HSC | ex vivo | [109] |
| DMD | Dystrophin | CRISPR and TALEN | NHEJ-mediated removal of stop codon, splice site or frameshifted exon. HR-based repair of Dystrophin gene | in vivo | [81,87–89] |
| alpha-hemoglobinopathies | BCL11A | TALEN | NHEJ-based deletion of erythroid-specific enhancer in BCL11A gene | ex vivo | [85] |
| beta-globin | CRISPR and TALEN | HR-based repair of sickle cell anemia mutation in iPSCs and differentiate to erythrocytes | ex vivo | [117, 118] | |
| alpha-tyrosinemia | Fah | CRISPR | HR-based repair of Fah gene in liver cells | in vivo | [132] |
| Hypercholesterolemia | PCSK9 | CRISPR | NHEJ-based deletion of PCSK9 gene in liver cells | in vivo | [53,133] |
| Infectious disease | |||||
| Hepatitis B virus | Viral | CRISPR and TALEN | NHEJ-induced frameshifts in essential HBV genes | in vivo | [9–14] |
| HIV | CCR5 | ZnF and CRISPR | NHEJ-induced frameshifts in CCR5 gene in CD4+ T-cells to prevent viral spread | ex vivo | [74–78] |
| Viral LTR | CRISPR | NHEJ-based deletion of integrated virus | ex vivo | [17–19] | |
| Staphylococcus aureus | Bacterial | CRISPR | NHEJ-induced frameshifts in essential S. aureus genes or antibiotic resistance genes | in vivo | [15] |
| Galleria mellonella | Bacterial | CRISPR | NHEJ-induced frameshifts in essential G. mellonella genes. | in vivo | [16] |
Non-homologous end joining
Repair of a DSB by NHEJ essentially involves re-ligation of the DNA ends, but is somewhat error prone and occasionally small insertions or deletions (indels) of a few bases can occur at the cut site. These indels are typically used to generate frameshift mutations in protein coding sequences resulting in loss of protein function (Figure 2). This has been applied to bacterial pathogens such as Galleria mellonella or antibiotic resistant S. aureus, where CRISPR nucleases targeting the bacterial genome or antibiotic resistance genes can block bacterial spread, and enable loss of antibiotic resistance [15,16]. Additionally, CRISPR/Cas9 or TALENs targeting key open reading frames in the genome of hepatitis B virus have also been shown to be effective in blocking viral protein expression and replication [9–14].
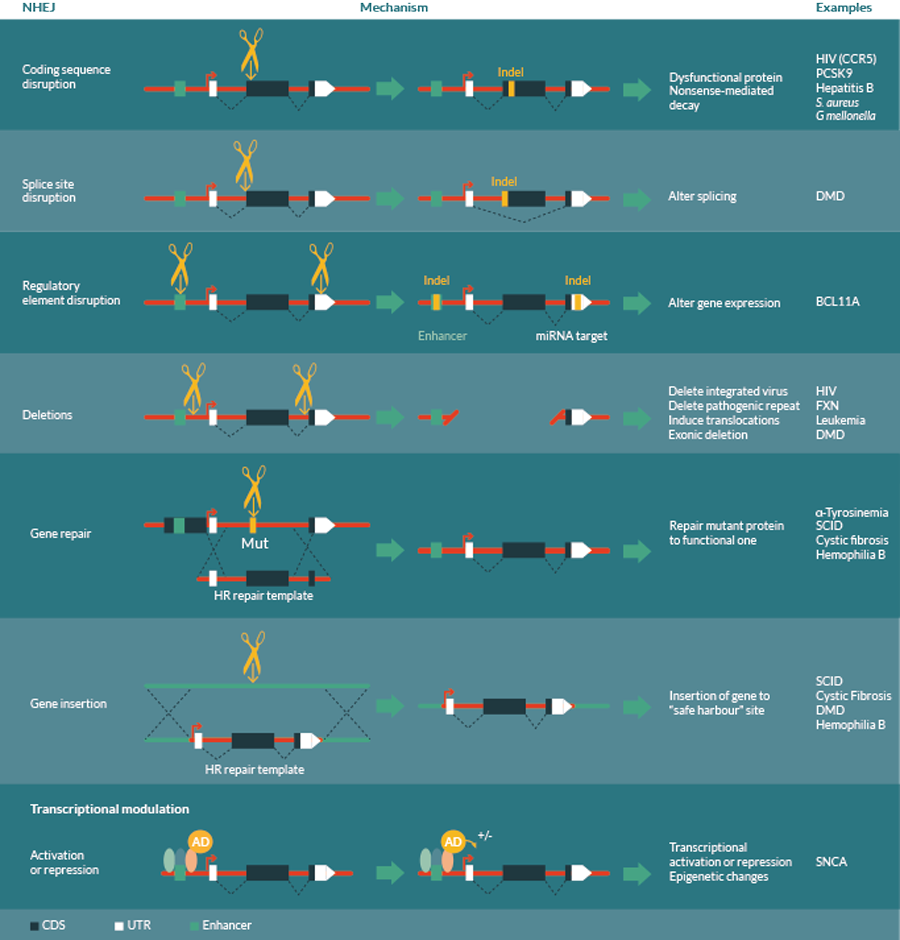
The choice of strategy depends on the nature of the disease, and may include non-homologous end joining (NHEJ) based mutations, homologous recombination (HR) based repair or transcriptional modulation by activation or repression domain fusions. NHEJ based mutations may be used to generate coding sequence disruptions to inactivate genes, splice site disruption to alter splicing patterns, enhancer or other regulatory element disruption to alter gene expression or deletions between two cuts to remove genomic regions. HR-based repair can be used to accurately repair mutations in genes, or insert transgenes into a defined “safe harbor” sites to avoid problems with random insertion. Transcriptional modulation may include activation or repression of transcription, or epigenetic modifications to cause semi-permanent changes to gene expression.
NHEJ-based loss of function mutations can also be used to introduce protective alleles that have been identified to confer resistance to particular diseases. For example, it was noted that individuals homozygous mutant for the HIV coreceptor gene, CCR5, were highly resistant to infection with HIV and yet otherwise healthy [74]. This has resulted in phase I trials using ZFNs to knockout the CCR5 gene in CD4+ T cells as a therapy for HIV infection [75–78].
However, indels generated by inefficient NHEJ can also be used to mutate other small functional regions of the genome (Figure 2). These may include splice sites to alter splicing patterns, micro RNA (miRNA) target sites or miRNA genes themselves to alter post-transcriptional regulatory networks [79], or transcription factor binding sites to alter gene expression profiles of specific genes [80]. For instance, restoration of Dystrophin function, the causative lesion in Duchenne’s muscular dystrophy (DMD), can be achieved by skipping of exons that contain frameshifting mutations. CRISPR/Cas9 reagents have been targeted to these splice site regions and been shown to restore gene function [81]. Interestingly, most disease-linked single nucleotide polymorphisms (SNPs) lie outside of protein-coding sequence (93%, Encyclopaedia of DNA elements [ENCODE] [82]), and many of them fall within gene regulatory elements as defined by DNAse hypersensitivity (perhaps more than 50% [83]) suggesting that this set of targets may be able to revert disease phenotypes [83,84]. Importantly, these regulatory sequences can have tissue-specific or developmental stage dependent effects on gene expression, making them good candidates for therapeutic intervention, such as those near the BCL11A gene involved in b hemoglobinopathies [80,85]. Mutation of these regulatory elements specifically reduces gene expression in erythroid but not B-lymphoid cells, allowing specific reactivation of fetal hemoglobin (HbF) in erythroid lineages, but leaving the non-erythroid functions of BCL11A unaffected [80,85].
Interestingly, pairs of DSBs can also result in deletion and inversion of the intervening sequence, or even translocations between chromosomes, making it possible to induce larger chromosomal deletions or rearrangements (Figure 2). This has applications in deleting genes or repeat expansions present in for example Friedreichs Ataxia [86], removal of exons in for example DMD [81,87–89], or reverting chromosomal translocations observed in certain leukemias [90–92]. Interestingly, such large deletions can be used to target latent viral infections such as HIV that are currently difficult or impossible to treat. CRISPR/Cas9 nucleases targeted to the long terminal repeats (LTR) of the virus are able to recognize cells with a viral integration, excising the viral genome and generating cells free of integrated virus [17–19] (Table 1).
Homologous recombination
HR-based repair systems normally use the homologous region of the sister chromatid as a template for high fidelity repair, and therefore only occur after DNA replication, in late S and G2 phases of the cell cycle. By supplying an excess of a desired repair template, this mechanism can be exploited to enable precise, directed changes to be achieved such as the repair of pathogenic mutations, deletion of defined regions of the DNA or integration of transgenes to a specific genomic location [73] (Figure 2, Table 1). Expression of transgenes from a defined “safe harbor” site can prevent problems with undesired mutations generated by random insertions, and ensure uniform expression levels. Recently, ZFNs have been used to integrate Factor VIII and IX transgenes into the albumin locus to restore functional gene expression in mouse models of hemophilia [93], providing an example of such a strategy.
It is highly beneficial in many therapeutic contexts to make very precise changes to the DNA sequence, and indeed essential in certain cases. Many genetic diseases such as cystic fibrosis, SCID, hemophilia B and a-tyrosinemia require the repair of mutant alleles in a precise manner to reverse point mutations that are causative in disease. However, HR-based repair is currently difficult to implement at high efficiency and requires simultaneous delivery of an appropriate repair template. Importantly, NHEJ repair vastly predominates over HR in most cell types, especially those which have withdrawn from the cell cycle, such as neurons, where the latter repair process is absent. Significant steps have been made to improve the rates of HR by inhibiting NHEJ pathways both chemically and with transient knockdown by RNAi [94,95], manipulating the timing of genome engineering relative to the cell cycle [96], use of ssDNA templates for homology-directed repair [97] or chemical enhancement of HR pathways [98]. There is little doubt that the rates will be improved still further with additional developments of these systems. Interestingly, a third type of DNA repair, microhomology-mediated end joining has emerged as a useful, if less precise alternative to HR that occurs at different phases of the cell cycle, does not require the HR repair machinery and therefore could be applied to different cell types [99].
Transcriptional modulation
Although introduction of changes in the DNA sequence are beneficial in many ways, such changes are permanent, which brings additional risks should there be undesired effects, or off-targeting. With this in mind, it is also possible to recruit other functional protein domains to desired sites in the genome using these site-specific DNA-binding factors. This can elicit transient up- or down-regulation of the transcriptional activity of specific genes or even complex alterations to transcriptional profiles [58,100–105]. This strategy could be used to restore gene expression patterns for diseases where these are perturbed, such as reverting the overexpression of the α-synuclein (SNCA) gene that often occurs in Parkinson’s disease [106]. It is also possible to recruit chromatin-modifying factors to specific sites to modulate the epigenetic status of the cell, a feature which is often perturbed in disease [107,108]. Interestingly, once certain epigenetic states are altered, they are able to persist after removal of the initiating factor, thus providing a semi-permanent change to the cell without altering DNA sequence.
Delivery systems
Application of therapeutic genome engineering requires delivery of the reagents to cells, which can be achieved either directly in vivo, or to patient-derived cells cultured ex vivo, that are subsequently re-implanted. Each of these has its own advantages, and the choice of system will depend on the specific disease, the accessibility of tissue and availability of in vitro culture systems (Table 1). Also, certain diseases are inherently easier to target, since either they only require a small number of repaired cells in order to rescue the pathological symptoms, or the repaired cells obtain a growth advantage compared to the diseased cells, and therefore will eventually outcompete them when grafted in vivo.
Ex vivo
Certain tissues, notably within the hematopoietic system, can be removed from patients, cultured in vitro and re-implanted after genetic manipulation. However, the number of situations amenable to such manipulation is limited, often due to inaccessibility of the tissue or lack of appropriate in vitro culture conditions. However, when this is possible, it provides significant advantages for genome engineering, namely in the delivery of constructs, and the selection of correctly repaired cells prior to implantation. This is particularly relevant when the efficiency of modification is low, such as for HR-based repair strategies, since those cells with the desired modifications can be clonally selected from a small proportion of modified cells and fully analyzed prior to re-implantation.
This can be a tremendously useful strategy for diseases of the hematopoietic system such as SCID [109] and HIV [75], where cells can be removed, cultured, manipulated and transplanted into patients. In the case of HIV treatment, there is also a fitness disadvantage of HIV infection in CD4+ T-cells, which is highly beneficial during re-implantation of modified cells. Deletion of the CCR5 co-receptor gene not only prevents viral infection, but these modified cells will outcompete the remaining HIV-infected cells in the patient [75–78]. Gene correction has also been shown to be possible at the IL2RG locus, where hematopoietic stem cells (HSCs) from a patient with SCID-X1 were repaired by an HR-based mechanism [109]. Given the corrected cells have a selective advantage over the mutant cells, these HSCs can be produced in sufficient quantities for re-implantation. HSCs were able to be autologously transplanted into mice and gave rise to essentially all cell types in the hematopoietic lineage [109], making this strategy extremely valuable for the treatment of many other hematological disorders [110].
Certain other adult stem cells such as intestinal organoids [111] can be cultured in vitro, manipulated genetically and effectively grafted into the colon of mice carrying chemically induced mucosal lesions [112]. A recent study of organoids derived from cystic fibrosis patients has demonstrated correction of the causative mutation in the CFTR gene [113], and reverts the cellular phenotypes caused by this mutation. This therefore provides an opportunity to treat the intestinal phenotypes associated with this disease.
These studies provide important paradigms for further development in this area. Indeed, the advent of induced pluripotent stem cells (iPSCs) [114,115] and their ability to be differentiated into essentially any cell type vastly increases the general applicability of such strategies [116]. A recent report has shown that it is possible to use CRISPR- or TALEN-directed HR in iPSCs to correct the mutation in the b-globin gene that causes sickle cell anemia, and differentiate the resulting cells into disease-free erythrocytes [117,118]. However, ex vivo genetic engineering is still limited by the inefficient differentiation into certain adult cell types, and problems with graft rejection upon re-implantation [116,119]. Equally, certain tissues such as the brain are relatively inaccessible and there are inherent difficulties in re-implanting new cells due to the complex tissue architectures involved, limiting its use in treating certain diseases.
In vivo
In vivo delivery of genome editing components is an attractive alternative that circumvents the problems of tissue accessibility, ability to culture cells in vitro and issues with re-implantation. However, delivery of the components necessary for genome engineering is fraught with the same problems as other gene therapy strategies, predominantly in terms of the efficiency of cellular uptake, and immune responses to such interventions [120–122]. Importantly, the genetic changes made by genome engineering are inherently heterogeneous in nature, introducing cell-to-cell variability in genotype, and are delivered to many different cell types throughout the body. This can be an advantage in some situations where the pathology results from multiple tissues, but may also create additional complications through the generation of genetic changes in otherwise healthy tissues.
A major advantage of genome engineering for gene therapy applications is that the genetic changes that are created are permanent, so they only need to be generated once per cell. Indeed this could be achieved by multiple sequential treatments that could be administered over a period of time, each of which only targets a small proportion of cells. Additionally, if the genetic changes can be made in stem cells, these will repopulate the tissue with the repaired genome, and therefore offer a permanent solution.
Genome engineering reagents can be supplied in a number of different ways – as DNA plasmids, incorporation into viral vectors, or provided as in vitro-transcribed mRNAs/sgRNAs or recombinant proteins. Each strategy is being developed, and has advantages for gene therapy in certain situations, or in delivery to certain tissues [121–124].
Viral vectors are perhaps the most advanced of these, and efficient systems of delivery of lentiviral (LV) [123] and adenovirus-associated virus (AAV) [124] vectors to a variety of different tissues have been developed and successfully used to deliver ZFNs in vivo [97]. However, both TALEN and CRISPR/Cas9 systems require expression of relatively large proteins. For example, the S. pyogenes Cas9 protein is too large to be packaged into a single AAV vector with its cognate sgRNA and it is not possible to package a TALEN pair in a single AAV vector (Figure 1). Development of such systems is ongoing, and recent advances include use of dual AAV vectors for CRISPR/Cas9 delivery [125], and development of an AAV vector for the smaller S. aureus Cas9 protein [53]. This is able to be packaged into a single AAV along with its respective sgRNA, and has been shown to be highly effective in vivo in mice [53]. Other smaller Cas9 variants such as the Cpf1 protein from Francisella novicida provide opportunities for other analogous systems [55]. Many current viral vectors will integrate randomly into the genome, and this has caused problems in previous gene therapy trials [3]. The fact that genome engineering reagents only need to be transiently expressed to cause permanent effects on the genome raises the possibility to use integration-deficient forms of the viral vectors removing this caveat [109].
Equally, systems for direct delivery of DNA, RNA and protein are becoming available, including liposome-, nanoparticle- and peptide-mediated delivery systems that are highly effective in certain tissues [121,122,126]. These can be complexed with the appropriate biological molecule non-covalently or covalently [127–129], and significant progress is being made in the stability of such complexes in serum, and their delivery into the cell cytoplasm [121]. Similarly, exosomal vesicles have shown promise as delivery systems for many tissues, and can be targeted to specific cell types by decoration with particular cell surface markers [130]. These delivery systems have the advantage that they will only express transiently, reducing the risks of off-target effects. In the case of mRNA and protein they are also unable to integrate non-specifically into the genome, eliminating such unintended side-effects.
Such in vivo delivery is especially applicable when the disease only requires a small proportion of cells to be repaired in order to relieve symptoms, such as in the case of hemophilia B or α-tyrosinemia. Hemophilia B results from a defect in Factor IX activity, that results in defects in blood clotting. Restoration of its activity to even a few percent of normal levels in the liver can transform the pathological symptoms of the patient [131]. A study has used AAV vectors to deliver ZFNs and an homologous repair template, resulting in correction of up to 7% of alleles in mouse liver, and an amelioration in the associated phenotypes [97]. Similarly, a mouse model of hereditary α-tyrosinemia resulting from a defect in splicing of the Fah gene can be corrected by hydrodynamic delivery of CRISPR/Cas9 nucleases and an oligonucleotide repair template [132]. Although the initial correction is only in 0.4% of liver cells, these gain a selective advantage and expand to form around 33% of liver tissue after 30 days, resulting in correction of the pathogenic phenotypes [132].
High efficiencies of mutagenesis of up to 50% have been achieved in murine liver cells in vivo using adenoviral vectors to deliver CRISPR/Cas9 to generate NHEJ-mediated loss of function mutations in the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene [133]. This results in a 35–40% reduction in blood cholesterol levels, and may have therapeutic potential for prevention of cardiovascular disease.
Conclusions & future challenges
The application of genome engineering approaches to human genetic disease is an exciting prospect, with the potential to correct disease-causing aberrations and result in a permanent cure. The ease with which this can be achieved and the optimal strategies involved depends strongly on the nature of the disease. Diseases where a partial restoration of function is sufficient to rescue the pathology, such as cystic fibrosis, or hemophilia B, or where wild-type cells have a selective advantage in vivo such as SCID-X1, HIV or α-tyrosinemia will initially provide the most attractive candidates for treatment. Equally, although considerable advances have been made in understanding the genetic basis of complex heritable polygenic disorders through analyses such as genome-wide association studies, few examples show large effects of a single allelic variant [134]. Therefore at least initially, monogenic disorders will provide the best chances for therapeutic genetic intervention.
Loss of function mutations generated by NHEJ repair are the most efficient and easiest to introduce but there are a limited number of diseases that can be treated in this manner, such as bacterial or viral infections, those where protective null alleles have been identified, or where splicing, transcription or miRNAs can be modulated. Therefore, an important future challenge will be to increase the rates of HR-based repair to a level where they are useful in vivo. Although significant progress has been made along these lines, further improvements will be necessary before this is generally applicable, other than to those diseases where repair in a small proportion of cells is sufficient. Similarly, the efficiency of delivery systems and their temporal and tissue specificity also needs to be addressed in order for such in vivo therapies to become a reality.
Equally important for in vivo gene therapies is the identification and elimination of off-target effects, which may cause unintended and permanent changes elsewhere in the genome. Whilst these problems have been addressed to a large extent in the research environment, this is particularly relevant when therapies are applied to the entire body, with approximately 3.7 x 1013 cells [135]. In this context, even an extremely small proportion of off-target mutagenesis could become relevant.
Another promising avenue that removes some of the problems of efficiency and off targeting is ex vivo manipulation of patient-derived cells to obtain the correct genetic changes. The ability to characterize both the desired and off-target changes in the genome would allow delicate and precise manipulations to be performed, and reduce the chances of undesirable effects. Further work to improve culture systems for adult stem cells, and techniques for their efficient re-implantation will therefore be important if this strategy is to be more generally employed.
Perhaps the most extreme and controversial example of ex vivo genome engineering is the modification of the germline, resulting in every cell in the body containing the desired changes. This has been extremely successful in the case of mice [49] and even monkeys [46], and recent work has suggested that this is possible in humans [136]. However, there are many ethical implications to such modifications, not least of which being that any effects, beneficial or detrimental, will be passed on to subsequent generations, therefore irreversibly affecting the course of human evolution [7,8].
The use of genome engineering for therapeutic applications is a stimulating, fast moving field and perhaps one of the most exciting aspects of such therapies is their general applicability. Once the systems for modification of the genome, and delivery of the reagents and are established, it can be applied with relatively minor modifications to essentially any genetic disease. The developments in this area in the coming years will no doubt be extremely interesting, and may signify the beginnings of a cure for many genetic diseases.
Acknowledgements
This work was supported by the John Fell OUP Research Fund, and the Wellcome Trust Institutional Strategic Support Fund, and the Departments of Pathology, Biochemistry, Pharmacology and Physiology Anatomy and Genetics at the University of Oxford. The author would like to thank Dr Sarah Cooper for critical evaluation of the manuscript.
Financial disclosure
The author has no relevant financial involvement with an organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock options or ownership, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.
References
1.De Fougerolles AR. Delivery vehicles for small interfering RNA in vivo. Hum. Gene Ther.. 2008; 125–132. CrossRef
2.Kay MA. State-of-the-art gene-based therapies: the road ahead. Nat. Rev. Genet. 2011; 316–328. CrossRef
3.Check E. A tragic setback. Nature. 2002; 116–118. CrossRef
4.Urnov FD, Rebar EJ, Holmes MC, Zhang HS & Gregory PD. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010; 636–646.CrossRef
5.Bogdanove AJ & Voytas DF. TAL effectors: customizable proteins for DNA targeting. Science 2011; 1843–1846. CrossRef
6.Hsu PD, Lander ES & Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014; 1262–1278. CrossRef
7.Bosley KS, Botchan M, Bredenoord AL et al. CRISPR germline engineering–the community speaks. Nat. Biotechnol. 2015; 478–486. CrossRef
8.Lanphier E, Urnov F, Haecker SE, Werner M & Smolenski J. Don’t edit the human germ line. Nature. 2015; 410–411. CrossRef
9.Dong C, Qu L, Wang H et al. Targeting hepatitis B virus cccDNA by CRISPR/Cas9 nuclease efficiently inhibits viral replication. Antiviral Res. 2015; 110–117. CrossRef
10.Kennedy EM, Bassit LC, Mueller H et al. Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISPR/Cas RNA-guided DNA endonuclease. Virology 2015; 196–205. CrossRef
11.Lin SR, Yang HC, Kuo YT et al. The CRISPR/Cas9 System Facilitates Clearance of the Intrahepatic HBV Templates in vivo. Mol. Ther. Nucleic Acids 2014; e186. CrossRef
12.Ramanan V, Shlomai A, Cox DB et al. CRISPR/Cas9 cleavage of viral DNA efficiently suppresses hepatitis B virus. Sci. Rep. 2015; 10833.CrossRef
13.Zhen S, Hua L, Liu YH et al. Harnessing the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated Cas9 system to disrupt the hepatitis B virus. Gene Ther. 2015; 404–12. CrossRef
14.Bloom K, Ely A, Mussolino C, Cathomen T & Arbuthnot P. Inactivation of hepatitis B virus replication in cultured cells and in vivo with engineered transcription activator-like effector nucleases. Mol. Ther. 2013; 1889–97. CrossRef
15.Bikard D, Euler CW, Jiang W et al. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol. 2014; 1146–1150. CrossRef
16.Citorik RJ, Mimee M & Lu TK. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol. 2014; 1141–1145. CrossRef
17.Ebina H, Misawa N, Kanemura Y & Koyanagi Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013; 2510. CrossRef
18.Hu W, Kaminski R, Yang F et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl Acad. Sci. USA 2014; 11461–1146. CrossRef
19.Liao HK, Gu Y, Diaz A et al. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat. Commun. 2015; 6413. CrossRef
20.Choo Y & Klug A. Selection of DNA binding sites for zinc fingers using rationally randomized DNA reveals coded interactions. Proc. Natl Acad. Sci. USA 1994; 11168–11172. CrossRef
21.Choo Y & Klug A. Toward a code for the interactions of zinc fingers with DNA: selection of randomized fingers displayed on phage. Proc. Natl Acad. Sci. USA 1994; 11163–11167. CrossRef
22.Del Rio S, Menezes SR & Setzer DR. The function of individual zinc fingers in sequence-specific DNA recognition by transcription factor IIIA. J. Mol. Biol. 1993; 567–579. CrossRef
23.Bulyk ML, Johnson PL & Church GM. Nucleotides of transcription factor binding sites exert interdependent effects on the binding affinities of transcription factors. Nucleic Acids Res. 2002; 1255–12561. CrossRef
24.Pabo CO, Peisach E & Grant RA. Design and selection of novel Cys2His2 zinc finger proteins. Annu. Rev. Biochem. 2001; 313–340. CrossRef
25.Kim YG, Cha J & Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl Acad. Sci. USA 1996; 1156–1160. CrossRef
26.Miller JC, Holmes MC, Wang J et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat. Biotechnol. 2007; 778–785.CrossRef
27.Szczepek M, Brondani V, Buchel J et al. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat. Biotechnol. 2007; 786–793. CrossRef
28.Boch J, Scholze H, Schornack S et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009; 1509–12. CrossRef
29.Bonas U, Stall RE & Staskawicz B. Genetic and structural characterization of the avirulence gene avrBs3 from Xanthomonas campestris pv. vesicatoria. Mol. Gen. Genet. 1989; 127–136. CrossRef
30.Moscou MJ & Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science 2009; 1501. CrossRef
31.Cermak T, Doyle EL, Christian M et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011; e82. CrossRef
32.Li T, Huang S, Zhao X et al. Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res. 2011; 6315–6325. CrossRef
33.Reyon D, Tsai SQ, Khayter C et al. FLASH assembly of TALENs for high-throughput genome editing. Nat. Biotechnol. 2012; 460–465. CrossRef
34.Sanjana NE, Cong L, Zhou Y et al. A transcription activator-like effector toolbox for genome engineering. Nat. Protoc. 2012; 171–192. CrossRef
35.Cade L, Reyon D, Hwang WY et al. Highly efficient generation of heritable zebrafish gene mutations using homo- and heterodimeric TALENs. Nucleic Acids Res. 2012; 8001–8010. CrossRef
36.Ishino Y, Shinagawa H, Makino K, Amemura M & Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987; 5429–5433.
37.Jansen R, Embden JD, Gaastra W & Schouls LM. Identification of genes that are associated with DNA repeats in prokaryotes.Mol. Microbiol. 2002; 1565–1575. CrossRef
38.Barrangou R, Fremaux C, Deveau H et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007; 1709–1712. CrossRef
39.Garneau JE, Dupuis ME, Villion M et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010; 67–71. CrossRef
40.Brouns SJ, Jore MM, Lundgren M et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 2008; 960–964. CrossRef
41.Gasiunas G, Barrangou R, Horvath P & Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl Acad. Sci. USA 2012; E2579–2586. CrossRef
42.Jinek M, Chylinski K, Fonfara I et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012; 816–821. CrossRef
43.Cong L, Ran FA, Cox D et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013; 819–823. CrossRef
44.Jinek M, East A, Cheng A et al. RNA-programmed genome editing in human cells. Elife 2013; e00471. CrossRef
45.Mali P, Yang L, Esvelt KM et al. RNA-guided human genome engineering via Cas9. Science 2013; 823–826. CrossRef
46.Niu Y, Shen B, Cui Y et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 2014; 836–843. CrossRef
47.Bassett AR, Tibbit C, Ponting CP & Liu JL. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep. 2013; 220–228. CrossRef
48.Reyes LM, Estrada JL, Wang ZY et al. Creating class I MHC-null pigs using guide RNA and the Cas9 endonuclease. J. Immunol. 2014; 5751–5757. CrossRef
49.Wang H, Yang H, Shivalila CS et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013; 910–918. CrossRef
50.Shalem O, Sanjana NE, Hartenian E et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014; 84–87. CrossRef
51.Wang T, Wei JJ, Sabatini DM & Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014; 80–84. CrossRef
52.Anders C, Niewoehner O, Duerst A & Jinek M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014; 569–573. CrossRef
53.Ran FA, Cong L, Yan WX et al. in vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015; 186–191. CrossRef
54.Hou Z, Zhang Y, Propson NE et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc. Natl Acad. Sci. USA 2013; 15644–15649. CrossRef
55.Zetsche B, Gootenberg JS, Abudayyeh OO et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015; 63(3), 759–771. CrossRef
56.Kleinstiver BP, Prew MS, Tsai SQ et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015; 481–485. CrossRef
57.Fu Y, Foden JA, Khayter C et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013; 822–826. CrossRef
58.Mali P, Aach J, Stranges PB et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013; 833–838. CrossRef
59.Tsai SQ, Zheng Z, Nguyen NT et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015; 187–197.
60.Pattanayak V, Lin S, Guilinger JP et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013; 839–843. CrossRef
61.Smith C, Gore A, Yan W et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell 2014; 12–13. CrossRef
62.Veres A, Gosis BS, Ding Q et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell 2014; 27–30. CrossRef
63.Hsu PD, Scott DA, Weinstein JA et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013; 827–832. CrossRef
64.Frock RL, Hu J, Meyers RM et al. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 2015; 179–186.
65.Wang X, Wang Y, Wu X et al. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat. Biotechnol. 2015; 175–178. CrossRef
66.Ran FA, Hsu PD, Lin CY et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013; 1380–1389. CrossRef
67.Tsai SQ, Wyvekens N, Khayter C et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat. Biotechnol. 2014; 569–576. CrossRef
68.Fu Y, Sander JD, Reyon D, Cascio VM & Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014; 279–284. CrossRef
69.Fu Y, Reyon D & Joung JK. Targeted genome editing in human cells using CRISPR/Cas nucleases and truncated guide RNAs. Methods Enzymol. 2014; 21–45. CrossRef
70.Nishimasu H, Cong L, Yan WX et al. Crystal Structure of Staphylococcus aureus Cas9. Cell 2015; 1113–1126. CrossRef
71.Nishimasu H, Ran FA, Hsu PD et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014; 935–949. CrossRef
72.Shrivastav M, De Haro LP & Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008; 134–147. CrossRef
73.Bibikova M, Beumer K, Trautman JK & Carroll D. Enhancing gene targeting with designed zinc finger nucleases. Science 2003; 764. CrossRef
74.Liu R, Paxton WA, Choe S et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996; 367–377. CrossRef
75.Tebas P, Stein D, Tang WW et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014; 901–10. CrossRef
76.Holt N, Wang J, Kim K et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat. Biotechnol. 2010; 839–847. CrossRef
77.Perez EE, Wang J, Miller JC et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 2008; 808–816. CrossRef
78.Ye L, Wang J, Beyer AI et al. Seamless modification of wild-type induced pluripotent stem cells to the natural CCR5Delta32 mutation confers resistance to HIV infection. Proc. Natl Acad. Sci. USA 2014; 9591–9596. CrossRef
79.Bassett AR, Azzam G, Wheatley L et al. Understanding functional miRNA–target interactions in vivo by site-specific genome engineering. Nature Commun. 2014; 4640. CrossRef
80.Canver MC, Smith EC, Sher F et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 2015; Sep 16. CrossRef
81.Li HL, Fujimoto N, Sasakawa N et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Reports 2015; 143–54. CrossRef
82.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 57–74.
83.Maurano MT, Humbert R, Rynes E et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012; 1190–1195. CrossRef
84.Pennisi E. The Biology of Genomes. Disease risk links to gene regulation. Science 2011; 1031. CrossRef
85.Bauer DE, Kamran SC, Lessard S et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 2013; 253–257. CrossRef
86.Pandolfo M. Friedreich ataxia. Arch. Neurol. 2008; 1296–1303. CrossRef
87.Ousterout DG, Kabadi AM, Thakore PI et al. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 2015; 6244. CrossRef
88.Long C, McAnally JR, Shelton JM et al. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 2014; 1184–1188. CrossRef
89.Ousterout DG, Perez-Pinera P, Thakore PI et al. Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol. Ther. 2013; 1718–1726. CrossRef
90.Blasco RB, Karaca E, Ambrogio C et al. Simple and rapid in vivo generation of chromosomal rearrangements using CRISPR/Cas9 technology. Cell Rep 2014; 1219–1227. CrossRef
91.Choi PS & Meyerson M. Targeted genomic rearrangements using CRISPR/Cas technology. Nat. Commun. 2014; 3728. CrossRef
92.Maddalo D, Manchado E, Concepcion CP et al. in vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 2014; 423–427. CrossRef
93.Sharma R, Anguela XM, Doyon Y et al. in vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood 2015; CrossRef
94.Chu VT, Weber T, Wefers B et al. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015; 543–548. CrossRef
95.Maruyama T, Dougan SK, Truttmann MC et al. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015; 538–542. CrossRef
96.Lin S, Staahl BT, Alla RK & Doudna JA. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 2014; e04766. CrossRef
97.Li H, Haurigot V, Doyon Y et al. in vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011; 217–221. CrossRef
98.Yu C, Liu Y, Ma T et al. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell 2015; 142–147. CrossRef
99.Nakade S, Tsubota T, Sakane Y et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat. Commun. 2014; 5560. CrossRef
100.Qi LS, Larson MH, Gilbert LA et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013; 1173–1183. CrossRef
101.Zalatan JG, Lee ME, Almeida R et al. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell 2015; 339–350. CrossRef
102.Cheng AW, Wang H, Yang H et al. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res 2013; 1163–1171. CrossRef
103.Farzadfard F, Perli SD & Lu TK. Tunable and multifunctional eukaryotic transcription factors based on CRISPR/Cas. ACS Synth. Biol. 2013; 604–613. CrossRef
104.Gilbert LA, Larson MH, Morsut L et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013; 442–51. CrossRef
105.Perez-Pinera P, Kocak DD, Vockley CM et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 2013; 973–976. CrossRef
106.Eschbach J & Danzer KM. alpha-Synuclein in Parkinson’s disease: pathogenic function and translation into animal models. Neurodegener. Dis. 2014; 1–17. CrossRef
107.Jakovcevski M & Akbarian S. Epigenetic mechanisms in neurological disease. Nat. Med. 2012; 1194–1204. CrossRef
108.Zhang Z & Zhang R. Epigenetics in autoimmune diseases: Pathogenesis and prospects for therapy. Autoimmun. Rev. 2015; CrossRef
109.Genovese P, Schiroli G, Escobar G et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014; 235–240. CrossRef
110.Weissman IL & Shizuru JA. The origins of the identification and isolation of hematopoietic stem cells, and their capability to induce donor-specific transplantation tolerance and treat autoimmune diseases. Blood 2008; 3543–53. CrossRef
111.Leushacke M & Barker N. Ex vivo culture of the intestinal epithelium: strategies and applications. Gut 2014; 1345–1354. CrossRef
112.Yui S, Nakamura T, Sato T et al. Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5(+) stem cell. Nat. Med. 2012; 618–23.CrossRef
113.Schwank G, Koo BK, Sasselli V et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013; 653–8. CrossRef
114.Takahashi K & Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 663–76. CrossRef
115.Okita K & Yamanaka S. Induced pluripotent stem cells: opportunities and challenges. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2011; 2198–2207. CrossRef
116.Schroeder IS. Stem cells: are we ready for therapy? Methods Mol. Biol. 2014; 3–21. CrossRef
117.Huang X, Wang Y, Yan W et al. Production of gene-corrected adult beta globin protein in human erythrocytes differentiated from patient IPSCs after genome editing of the sickle point mutation. Stem Cells 2015; 1470–1479. CrossRef
118.Voit RA, Hendel A, Pruett-Miller SM & Porteus MH. Nuclease-mediated gene editing by homologous recombination of the human globin locus. Nucleic Acids Res. 2014; 1365–1378. CrossRef
119.Boyd AS & Fairchild PJ. Approaches for immunological tolerance induction to stem cell-derived cell replacement therapies. Expert Rev Clin Immunol 2010; 435–48.CrossRef
120.Bessis N, GarciaCozar FJ & Boissier MC. Immune responses to Gene Ther.apy vectors: influence on vector function and effector mechanisms. Gene Ther. 2004; S10–S17. CrossRef
121.Yin H, Kanasty RL, Eltoukhy AA et al. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014; 541–555. CrossRef
122.Xu H, Li Z & Si J. Nanocarriers in Gene Ther.apy: a review. J Biomed Nanotechnol 2014; 3483–507. CrossRef
123.Rothe M, Modlich U & Schambach A. Biosafety challenges for use of lentiviral vectors in Gene Therapy. Curr Gene Ther. 2013; 453–468.
124.Kotterman MA & Schaffer DV. Engineering adeno-associated viruses for clinical Gene Therapy. Nat. Rev. Genet. 2014; 445–451. CrossRef
125.Swiech L, Heidenreich M, Banerjee A et al. in vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 2015; 102–106.
126.Andaloussi SE, Lehto T, Mager I et al. Design of a peptide-based vector PepFect6, for efficient delivery of siRNA in cell culture and systemically in vivo. Nucleic Acids Res. 2011; 3972–3987. CrossRef
127.Liu J, Gaj T, Patterson JT, Sirk SJ & Barbas CF, 3rd. Cell-penetrating peptide-mediated delivery of TALEN proteins via bioconjugation for genome engineering. PLoS One 2014; e85755. CrossRef
128.Ru R, Yao Y, Yu S et al. Targeted genome engineering in human induced pluripotent stem cells by penetrating TALENs. Cell Regen. (Lond.) 2013; 5. CrossRef
129.Ramakrishna S, Kwaku Dad AB, Beloor J et al. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res 2014; 1020–1027. CrossRef
130.Alvarez-Erviti L, Seow Y, Yin H et al. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011; 341–345. CrossRef
131.Lofqvist T, Nilsson IM, Berntorp E & Pettersson H. Haemophilia prophylaxis in young patients–a long-term follow-up. J. Intern. Med. 1997; 395–400. CrossRef
132.Yin H, Xue W, Chen S et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 2014; 551–553. CrossRef
133.Ding Q, Strong A, Patel KM et al. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ. Res. 2014; 488–492. CrossRef
134.Wray NR, Yang J, Hayes BJ et al. Pitfalls of predicting complex traits from SNPs. Nat. Rev. Genet. 2013; 507–515. CrossRef
135.Bianconi E, Piovesan A, Facchin F et al. An estimation of the number of cells in the human body. Ann. Hum. Biol. 2013; 463–471. CrossRef
136.Liang P, Xu Y, Zhang X et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell 2015; 363–372. CrossRef
Affiliation
Andrew Bassett
University of Oxford, Sir William Dunn School of Pathology, South Parks Road, Oxford, OX1 3RE
+44 (0)1865 285489
andrew.bassett@path.ox.ac.uk