Exciting developments in CRISPR/Cas9-mediated approaches for Duchenne MD
Cell Gene Therapy Insights 2015; 1(2), 215-230
10.18609/cgti.2015.022
The first use of the prokaryotic clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system in mammalian cells a couple of years ago paved the way for a revolution in the field of genome engineering. The availability of this simple-to-design, easy-to-use and multiplexing RNA-guided system enabled its widespread use in various applications. This technology has opened new avenues for the investigation and potential treatment of genetic diseases, such as Duchenne Muscular Dystrophy (DMD). This Expert Insight describes how CRISPR/Cas9 research could potentially be used therapeutically in the treatment of DMD, along with the principal hurdles and difficulties faced with its use, and hypothesizes on potential novel targets/uses of CRISPR/Cas9 in relation to DMD.
Submitted for Review: Sep 29 2015 Published: Dec 10 2015
Duchenne Muscular Dystrophy (DMD) is an hereditary, X-linked neuromuscular disease, resulting from mutations across the DMD gene. The subsequent absence of dystrophin protein prevents the correct formation of the dystrophin-associated protein complex (DAPC), a structural link between the intracellular actin and extracellular matrix. This compromises muscle stability and contractility, giving rise to progressive muscle wasting, the prominent clinical feature of this disease. Over time, muscle deterioration results in loss of ambulation, decline in respiratory and cardiac function and ultimately premature death between the second and third decade of life. Whilst the genetic basis of the disease is well established, care remains palliative and there is a clear unmet medical requirement for a gene therapy to address the underlying cause of this devastating disease [1]. A number of different gene therapies are currently in clinical trials and are reporting varying degrees of therapeutic benefit (reviewed in [2,3]). Briefly, these include adeno-associated virus (AAV) microdystrophin delivery [4], premature termination codon read-through using ataluren (TranslarnaTM) [5], exon-skipping [6–8] and utrophin upregulation [9,10]. These gene therapies would require repeat administration, and/or carry an adverse immunological risk, and/or are restricted by mutation specificity, all of which may limit their clinical relevance. Such problems may be circumvented through the use of genome engineering.
Therapeutic engineering of the DMD gene
Although the potential benefit of genome engineering strategies to correct the DMD gene is immense, this approach is challenged by the large size of the gene and the diversity of genetic mutations found in patients [11]. The mutation subsets and their relative occurrences are: large deletions 68%, duplications involving more than one exon 12%, and small mutations 20%, as reported by the Treat-NMD Global database [12]. These mutations do not occur uniformly across the length of the DMD gene; they are rather clustered into minor and major hotspots, between exons 2–20 and 45–55 respectively [3,13]. Genome editing could be used to affect permanent correction of numerous mutations via relatively few therapeutic strategies utilizing DNA repair mechanisms. This process orchestrates permanent changes in the mutated gene and, once corrected, the gene is expressed at its endogenous levels. Such correction of the primary genetic defect would prevent the necessity of repeated administration associated with traditional gene therapy approaches, and thus the likelihood of an adverse immunological reaction. Moreover, this can be used to target frequently occurring mutations or indeed intragenic mutational hotspots, excluding multiple exons and thus multiple mutations in the local region. The net result would produce a shortened functional dystrophin protein, whilst also increasing the therapeutic applicability of such an approach.
Genome engineering approaches classically depend upon the use of artificially engineered nucleases with the propensity to introduce a double strand break (DSB) at a defined location within the genome. Initially, this requirement was fulfilled by zinc finger nucleases (ZFNs), meganucleases (MGNs) and transcriptional activator-like effector nucleases (TALENs) [14–16]. ZFNs and TALENs became favored platforms, however they require two engineered protein motifs to confer specific binding, upstream and downstream of a DNA locus of interest, and the dimerization of FokI nucleases to cleave the DNA [17]. Although this represented a major advancement within the field, there were inherent limitations pertaining to the design of constructs being time consuming, expensive and resulting in variable efficacy. The subsequent discovery of CRISPR/Cas9 as a gene editing platform completely revolutionized the field due to the simplicity of guide design and its versatility [18,19].
Genome engineering is affected through the repair of the DSB. In the absence of a donor DNA template, repair via non-homologous end joining (NHEJ) will occur. This is an error prone method of DNA repair perpetuating small nucleotide insertions or deletions (INDELs), which has been shown to restore the reading frame and full-length or partial dystrophin protein expression [20,21] (Figure 1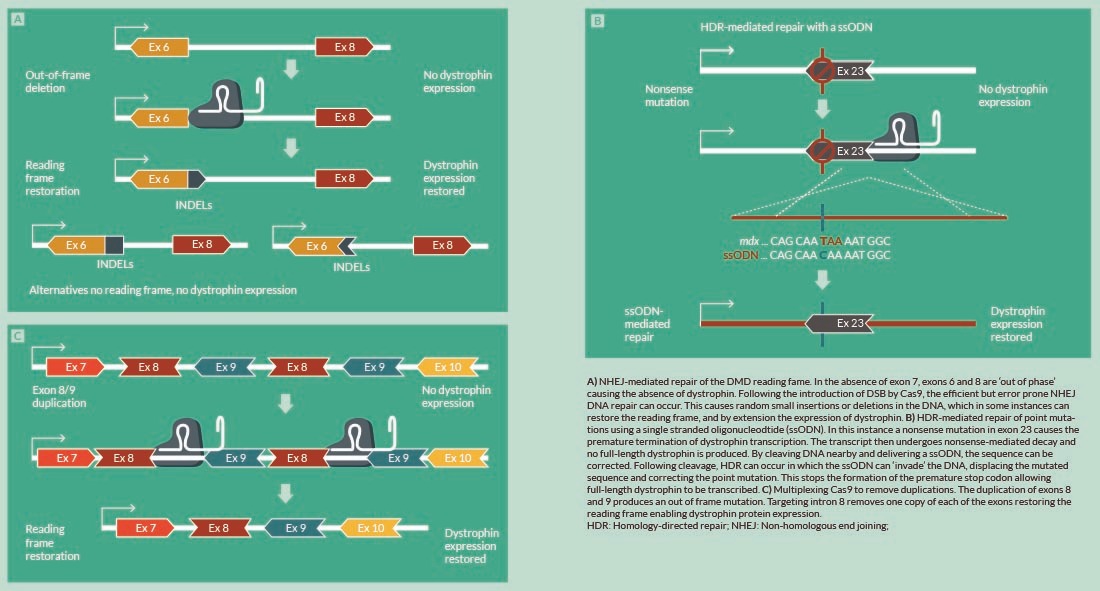
In contrast to NHEJ, HDR using ssODNs could specifically correct the DMD reading frame; insertion of the correct nucleotide(s) at the correct location will allow formation of the correct triplet codon and hence inclusion of the correct amino acid at the required position in the dystrophin protein. The delivery of extended pieces of coding DNA (cDNA) in the form of DNA donor repair templates have been used to address larger deletions. Our group previously demonstrated that a deletion of exons 45–52 was specifically reparable through use of a MGN and designed repair template [30] (Figure 2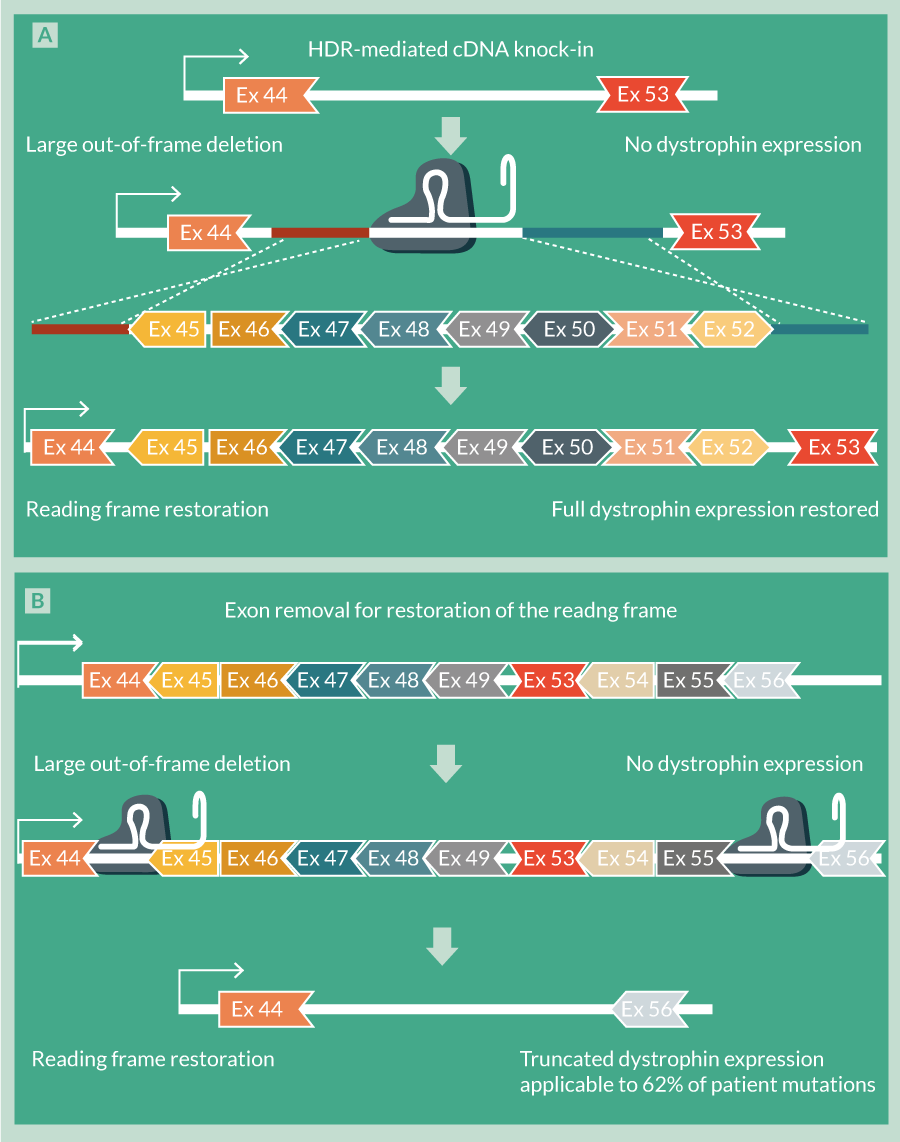
Careful selection of target site with consideration of the number of mismatches between gRNA and complementary sequence, their position and distribution is required [32–34].
- gRNAs should be designed to avoid poly-G, poly-C and poly-A rich sequences and where possible truncated gRNAs (17–18 bps) should be used since these have lower rates of off-target effects as a result of decreased mismatch tolerance [24]. gRNA secondary structure, which has the potential to prevent non-target interactions, and also chromatin levels of target should be taken into account [35]. Improved gRNA design tools are consistently being developed and should be exploited. The most recent design tools comparison reports that the sequence preference for CRISPR-mediated transcriptional regulation is substantially different from that for CRISPR/Cas9 knockout [36]. It has become increasingly apparent that Cas9 proteins can be engineered with higher specificity through alteration of their protospacer adjacent motif (PAM) dependence. For example, the recently described Neisseria meningitides Cas9 protein and Streptococcus thermophilus Cas9 proteins require PAMs with sequence 5’-NNNNGATT-3’ and 5’-NNAGAAW-3’, respectively offering much higher stringency in targeting than the established Streptococcus pyrogenes Cas9 (5’-NGG-3’) [18,37,38].
- Transfection conditions can have a dramatic effect on the level of off-target mutations as there is a clear relationship between gRNA and Cas9 concentration and undesired targeting [32,34].
- An examination of the reliability of existing techniques and the development of improved methodology for the detection of off-target mutations.
It should be noted that it has been reported that the use of paired nickases based on dead Cas9 (dCas9) as opposed to single wild-type Cas9 will increase target specificity by 1500 times [39,40], since off-target single nicks are faithfully repaired [41]. The fusion of FokI catalytic domain to dCas9 provides an additional layer of stringency since FokI has rigid spatial requirements for activity and will not cleave as a monomer [42,43].
For delivery, currently DNA (as plasmids expressing Cas9) and gRNA [44] and RNA [45,46] injection-based techniques are used for CRISPR/Cas9 delivery into organisms which is inefficient and non-specific. Efficiency of delivery would be increased through the use of vectors. To allow packaging of AAV vectors which are limited by the size of genetic cargo they can carry, smaller Cas9 orthologs from other microbes have recently been developed [47]. The use of the transfer RNA (tRNA) promoter expression strategy [48] could also greatly facilitate the construction of effective AAV-based Cas9/sgRNA vectors for future in vivo use. The use of AAVs to deliver CRISPR would allow the use of muscle-specific serotypes to enhance Cas9 and gRNA expression in muscle. Other vectors being exploited for CRISPR/Cas9 delivery include adenoviral [49–51] and lentiviral vectors [50,52]. Very recently, two groups have examined the potential of splitting the full wild-type Cas9 coding sequence between two AAV vectors to good effect [53,54]. The development of delivery systems that allow tissue- and cell-specific expression of CRISPR/Cas9 components has recently been demonstrated in zebrafish [55]. It is therefore hoped that muscle-specific expression is potentially possible. Other strategies to enhance delivery of CRISPR/Cas9 include the use of Cas9 recombinant protein [25] and cell-penetrating peptide (CPP) conjugates of this protein together with CPP–gRNA complexes [56]. Chemical alterations to synthesized gRNAs have recently been reported to enhance genome editing efficiency in human primary T-cells and CD34+ hematopoietic stem and progenitor cells without the toxicity associated with DNA delivery [57]. A novel delivery vehicle for CRISPR/Cas9 based on a biologically inspired yarn-like DNA nanoclew and synthesized by rolling circle amplification with palindromic sequences encoded to drive the self-assembly of nanoparticles has very recently been described [58]. To allow the precise temporal control of Cas9 gene expression, a light-inducible, user-defined, endogenous gene activation system based on CRISPR/Cas9 has been developed [59]. Application of these recent developments in optimizing delivery, targeting and expression to DMD gene editing should progress the work along the path to clinical readiness.
Strategies to tackle off-target effects, delivery problems as well as improvements in gRNA production and efficiency of HDR for full DMD gene repair are summarized in Table 1.
| Table 1 | ||
|---|---|---|
| Obstacle to overcome | Strategy to be used | Refs |
| Enhancement of HDR to fully repair the DMD gene | Use of a pair of nickases each containing a specific Cas9 mutant to produce single stranded cleavage which is preferentially repaired by HDR | [83,87,88] |
| Alteration of the synchronization of the cell cycle using reversible chemical inhibitors and controlled timing of CRISPR/Cas9 delivery | [26] | |
| Optimization of the homologous repair template e.g., longer arms of homology, use of ssODN | [26] | |
| Use of Cpf1-containing class 2 CRISPR system to introduce a staggered double strand break that could facilitate NHEJ-based cDNA insertion | [89] | |
| Reduction/elimination of off-target effects | Improved gRNA design | [32–34] |
| Avoidance of poly-G, poly-C and poly-A gRNA and use of truncated (17–18 bps) gRNAs | [24] | |
| Use of gRNA design software specific to type of genome engineering desired | [36] | |
| Use of engineered Cas9 proteins with more stringent PAM dependence e.g. Neisseria meningitides Cas9 protein and Streptococcus thermophilus Cas9 protein | [18,37,38] | |
| Optimization of transfection conditions since there is a clear gRNA and Cas9 concentration dependence on undesired targeting | [32,34] | |
| Improvement in the methodology used for the detection of off-target mutations | [90] | |
| Use of paired nickases to increase target specificity 1500-fold | [41] | |
| Fusion of FokI catalytic domain to nickases to provide an additional layer of stringency | [42,43] | |
| gRNA production | Use of the artificial gene RGR that undergoes self-catalyzed cleavage as an alternative to RNA polymerase III | [91] |
| Use of tissue/specific promoters as alternatives to the ubiquitously expressed U3 and U6 snRNA promoters | [91] | |
| Use of small, ∼70-bp tRNA promoters to express high levels of tRNA, sgRNA fusion transcripts that are efficiently and precisely cleaved by endogenous tRNase Z to release fully functional sgRNAs | [48] | |
| Delivery of CRISPR/Cas9 | Use of adenoviral and lentiviral vectors to increase efficiency and specificity | [49–52] |
| Use of smaller Cas9 orthologs from other microbes and tRNA promoter to allow packaging in AAV vectors | [47] | |
| Use of AAV serotypes that show skeletal muscle tropism | [92] | |
| Enhancement of delivery using Cas9 recombinant protein, CPP Cas9 conjugates and CPP–gRNA complexes | [25,56] | |
| Chemical alteration to synthesized sgRNA to enhance genome editing efficiency | [57] | |
| Use of novel CRISPR/Cas9 self-assembled delivery vehicle based on yarn-like DNA nanoclew | [58] | |
| Use of light-inducible, user-defined, endogenous gene activation system based on CRISPR/Cas9 to allow the precise temporal control of Cas9 gene expression | [59,63] | |
| AAV: Adeno-associated virus; CPP: Cell-penetrating peptide; gRNA: Guide RNA; PAM: Protospacer adjacent motif; sgRNA: Synthetic guide RNA; ssODN: Single strand DNA; tRNA: Transcriptional RNA. | ||
Generation of DMD animal models
The availability of emerging CRISPR-based gene editing strategies, with higher specificity and efficiency, has in turn impacted the availability and the development of animal models. In the DMD field, the mdx mouse is still the most widely known and used model since its discovery in the mid-’80s [60]. It carries a nonsense point mutation in exon 23 that causes dystrophin deficiency; however the disease has a milder and non-progressive course compared to the human counterpart. To achieve a closer clinical phenotype than that observed in the mdx mouse, large animals models are required [61]. The CRISPR/Cas9 system has been used for the generation of highly efficient, heritable, gene knockout in mice and rats, where the system has also been used to induce double-gene knockout with only a single microinjection of Cas9 and a mixture of sgRNAs into one-cell-stage rat embryos [62]. Importantly, CRISPR/Cas9 technology is also proven to be effective in non-traditional animal models, like goats and pigs, which are important for both agricultural and biomedical research and development [63–68]. This technology has facilitated simplified genetic modification [69] and creation of disease models [70] in non-human primates. Most recently, Chen and colleagues used CRISPR/Cas9 to target the monkey DMD gene to create mutations in order to recapitulate DMD. In their study, Cas9 induced mosaic mutations in over the 87% of the DMD alleles in muscle, which is also supported by the depletion of dystrophin protein in the targeted muscles and associated muscle pathology [70]. Interestingly, myostatin, a negative regulator of skeletal muscle mass, has also recently been targeted with Cas9/sgRNA by Zou Q et al, to produce a myostatin knock-out dog model [71]. Importantly, concerns arise due to the presence of mosaic mutations produced with the CRISPR system, which might confound the potential phenotype, especially since primates and large animals have much longer breeding times than rodents. Nonetheless, when developing DMD animal models, this aspect could turn out to be useful in acquiring deeper insights into the percentage of dystrophin-positive fibers required to restore the dystrophic phenotype.
This system has also been used for the generation of DMD-mutated rats [72], where the simultaneous targeting of DMD exon 3 and exon 16 resulted in the absence of dystrophin expression in the F0 generation. These mutations were heritable by the next generation, and F1 male rats exhibited similar phenotypes in their skeletal muscles. This demonstrates how two types of mutation apparent in human DMD can be relatively quickly recapitulated producing helpful platforms to explore potential repair strategies. The DMD rat shows a more marked decline in muscle strength, presence of degenerative/regenerative cycles in the heart and diaphragm and more marked muscle fibrosis; it could therefore serve as a superior model in terms of disease pathology to the mdx mouse that is currently so widely used for the testing of therapies for DMD.
The generation of the transgenic humanized mdx mouse model [73] has provided a valuable setting to test the in vivo efficacy of human-specific antisense oligonucleotides (AOs) prior to clinical trial [74–76]. This model expresses full-length human dystrophin protein on an mdx background. Use of an AO to induce exon skipping will disrupt the transcript reading-frame and thereby block dystrophin protein expression. The ideal animal model for testing AO efficacy would be one that is transgenic with human DMD gene carrying a relevant mutation. The use of CRISPR/Cas9 engineering of the humanized mdx mouse to generate such mouse models is yet to be executed but could provide improved translational development of therapies.
Animal models allow a means to investigate the efficacy of both ex vivo and in vivo genome engineering as a therapy for DMD. Yet to be performed, ex vivo genome-engineered stem cells could be engrafted onto dystrophic muscle and amelioration of disease phenotype assessed. In vivo genome correction of the mutation in the germline of the mdx mice using a designed sgRNA to target mdx exon 23 and an ssODN as a template for HDR-mediated gene repair has been reported [27]. The mdx zygotes were co-injected with Cas9 mRNA, sgRNA, and ssODN, and then implanted into pseudo-pregnant female mice. The “corrected” mdx progeny displayed from 2 to 100% correction of the mdx gene. Interestingly, the correction of only 17% of the mutant mdx alleles was sufficient to allow dystrophin expression in a majority of myofibers at a level of intensity comparable to that of wild-type mice, and the muscle exhibited fewer histo-pathologic hallmarks of muscular dystrophy than mdx muscle. Very recently, Xu et al have reported the excision of a 23-kb genomic region of the X-chromosome covering the mdx exon 23 mutation. In contrast to the previous approach described, this was performed using direct in vivo administration of CRISPR/Cas9 into post-natal mdx mice, but similarly resulted in truncated yet functional dystrophin protein being expressed [77].
Altogether these in vivo animal studies have begun to demonstrate a proof of concept that CRISPR/Cas9-based genome editing has the potential for pre-clinical development. Additionally, they demonstrate the potential to target specific tissues and regenerate transient models of disease without the breeding of engineered animals. The application of CRISPR/Cas9 technology for genome editing in a wide range of organisms will promote our understanding of disease pathology and provide animal models with therapeutic relevance for human diseases including DMD.
Unexplored potential of genome engineering for DMD
CRISPR/Cas9-based genome editing does not just hold potential for the correction of the DMD gene, but could also be used to address the multi-factorial nature of the disease using alternative targeting strategies. Cas9 does not necessarily have to be used to cleave DNA; an engineered Cas9 mutant with specific point mutations in its endonuclease domains such that it has no catalytic activity against DNA has been developed [78,79]. Rather than cleaving its target, this so called dCas9, upon binding to its target DNA upstream of a promoter through a gRNA, will prevent RNA polymerase binding and activity, and hence block transcription. It is therefore possible to use Cas9 as a modulator of transcription in a target-specific fashion. Since DMD is a multi-factorial disease in terms of the dystrophic, atrophic, fibrotic skeletal muscle phenotype, this dCas9-mediated transcriptional repression could be exploited. Cas9 can be used to address the dystrophin deficiency through NHEJ/HDR repair of the DMD gene as discussed above, and in its dCas9 mutated form can additionally be used to repress the transcription and hence expression of fibrogenic genes and negative regulators of muscle growth. Such an exciting strategy has not yet been explored with DMD, but could hold great therapeutic potential. It should be noted that the ability to target more than one gene with a single delivery vector has been made possible by the development of lentiviral [52,80] and AAV [81] vector systems for the modular delivery of multiple gRNAs.
Enhancement of gene expression is also possible using dCas9 through its tethering to transcriptional activators, such as VP64, the tetramer of the herpes simplex activation domain VP16 [78,82–84]. As a therapeutic strategy for DMD, VP64-tethered dCas9 could be used to activate the expression of genes involved in myogenesis, and could provide an exciting alternative to small molecule upregulation of utrophin [43], a dystrophin homolog.
For the regulation of expression of fibrogenic/anti-myogenic genes as potential adjunct therapies for DMD using CRISPR/Cas9, the targeting of RNA rather than DNA could hold distinct advantages. It could allow more specific repression/activation and potentially more efficient Cas9 binding. The identification of i) repression of a specific transcript by Francisella novicida Cas9 (FnCas9), guided by a small CRISPR/Cas-associated RNA [85] and ii) RNA targeting by a distinct CRISPR/Cas subtype (Type III) present in Pyrococcus furiosus [86] could provide the basis for programmable Cas9-mediated RNA interference of fibrotic/anti-myogenic transcripts.
With the appropriate gRNA design, CRISPR/Cas9 technology not only holds the potential to permanently modify all DMD mutations, irrespective of type, such that dystrophin protein expression is restored, but also to target associated dystrophic muscle phenotypes to provide an all-encompassing therapy. Application of recent developments in optimizing delivery, targeting and expression to DMD genome engineering should progress the work along the path to clinical readiness. The exponential growth of research resulting from the discovery of CRISPR/Cas9 may in the long term enable the effective cure of monogenic hereditary conditions including DMD, which previously appeared unachievable.
Financial & competing interests disclosure
The authors have no relevant financial involvement with an organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock options or ownership, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Acknowledgements
The authors gratefully acknowledge Muscular Dystrophy UK, Duchenne Parent Project and AFM-Telethon for their valued support of the gene editing work performed at Royal Holloway University of London.

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.
References
1.Bushby K, Finkel R, Birnkrant DJ et al. Diagnosis and management of Duchenne muscular dystrophy, part 1, diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010; 9(1), 77–93. CrossRef
2.Guiraud S, Chen H, Burns DT, Davies KE. Advances in genetic therapeutic strategies for Duchenne muscular dystrophy. Exp. Physiol. 2015 Jul 3; doi: 10.1113/EP085308 CrossRef
3.Jarmin S, Kymalainen H, Popplewell L, Dickson G. New developments in the use of gene therapy to treat Duchenne muscular dystrophy. Expert Opin. Biol. Ther. 2014; 14(2), 209–30. CrossRef
http://dx.doi.org/10.1517/14712598.2014.866087
4.Mendell JR, Campbell K, Rodino-Klapac L et al. Dystrophin immunity in Duchenne’s muscular dystrophy. New Engl. J Med. 2010; 363(15), 149–37. CrossRef
5.Bushby K, Finkel R, Wong B et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 2014; 50(4), 477–87. CrossRef
6.Mendell JR, Rodino-Klapac LR, Sahenk Z et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Annals Neurol. 2013; 74(5), 637–47. CrossRef
7.van Deutekom JC, Janson AA, Ginjaar IB et al. Local dystrophin restoration with antisense oligonucleotide PRO051. New Engl. J Med. 2007; 357(26), 2677–86. CrossRef
8.Voit T, Topaloglu H, Straub V et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 2014; 13(10), 987–96. CrossRef
9.Guiraud S, Squire SE, Edwards B et al. Second-generation compound for the modulation of utrophin in the therapy of DMD. Human Mol. Genetics 2015; 24(15), 4212–24. CrossRef
10.Tinsley JM, Fairclough RJ, Storer R et al. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PloS one 2011; 6(5), e19189. CrossRef
11.Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987; 50(3), 509–17. CrossRef
12.Bladen CL, Salgado D, Monges S et al. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Human Mutation 2015; 36(4), 395–402. CrossRef
13.Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006; 34(2), 135–44. CrossRef
14.Kay S, Hahn S, Marois E, Hause G, Bonas U. A bacterial effector acts as a plant transcription factor and induces a cell size regulator. Science 2007; 318(5850), 648–51. CrossRef
15.Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl Acad. Sci. USA 1996; 93(3), 1156–60. CrossRef
16.Li T, Huang S, Jiang WZ et al. TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Res. 2011; 39(1), 359–72. CrossRef
17.Bitinaite J, Wah DA, Aggarwal AK, Schildkraut I. FokI dimerization is required for DNA cleavage. Proc. Natl Acad. Sci. USA 1998; 95(18), 10570–5. CrossRef
18.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012; 337(6096), 816–21. CrossRef
19.Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. eLife 2013; 2, e00471. CrossRef
20.Li HL, Fujimoto N, Sasakawa N et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Reports 2015; 4(1), 143–54. CrossRef
21.Ousterout DG, Perez-Pinera P, Thakore PI et al. Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol. Therapy 2013; 21(9), 1718–26. CrossRef
22.Ousterout DG, Kabadi AM, Thakore PI, Majoros WH, Reddy TE, Gersbach CA. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 2015; 6, 6244. CrossRef
23.Ousterout DG, Kabadi AM, Thakore PI et al. Correction of dystrophin expression in cells from duchenne muscular dystrophy patients through genomic excision of exon 51 by zinc finger nucleases. Mol. Therapy 2015; 23(3), 523–32. CrossRef
24.Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nature Biotech. 2014; 32(3), 279–84. CrossRef
25.Kim S, Kim D, Cho SW, Kim J, Kim JS. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014; 24(6), 1012–9. CrossRef
26.Lin S, Staahl BT, Alla RK, Doudna JA. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife 2014; 3, e04766. CrossRef
27.Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, Olson EN. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 2014; 345(6201), 1184–8. CrossRef
28.Barton-Davis ER, Cordier L, Shoturma DI, Leland SE, Sweeney HL. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J. Clin. Invest.1999; 104(4), 375–81. CrossRef
29.Welch EM, Barton ER, Zhuo J et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007; 447(7140), 87–91. CrossRef
30.Popplewell L, Koo T, Leclerc X et al. Gene correction of a duchenne muscular dystrophy mutation by meganuclease-enhanced exon knock-in. Human Gene Ther. 2013; 24(7), 692–701. CrossRef
31.Heyer WD, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Ann. Rev Genetics 2010; 44, 113–39. CrossRef
32.Fu Y, Foden JA, Khayter C et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotech. 2013; 31(9), 822–6. CrossRef
33.Hsu PD, Scott DA, Weinstein JA et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotech. 2013; 31(9), 827–32. CrossRef
34.Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nature Biotech. 2013; 31(9), 839–43. CrossRef
35.Singh R, Kuscu C, Quinlan A, Qi Y, Adli M. Cas9-chromatin binding information enables more accurate CRISPR off-target prediction. Nucleic Acids Res. 2015; 43(18), e118. CrossRef
36.Xu H, Xiao T, Chen CH et al. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 2015; 25(8), 1147–57. CrossRef
37.Deveau H, Barrangou R, Garneau JE et al. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 2008; 190(4), 1390–400. CrossRef
38.Hou Z, Zhang Y, Propson NE et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc. Natl Acad. Sci. USA 2013; 110(39), 15644–9. CrossRef
39.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014; 346(6213), 1258096. CrossRef
40.Ran FA, Hsu PD, Lin CY et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013; 154(6), 1380–9. CrossRef
41.Ghezraoui H, Piganeau M, Renouf B et al. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol. Cell 2014; 55(6), 829–42. CrossRef
42.Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nature Biotech. 2014; 32(6), 577–82. CrossRef
43.Tsai SQ, Wyvekens N, Khayter C et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nature Biotech. 2014; 32(6), 569–76. CrossRef
44.Gratz SJ, Cummings AM, Nguyen JN et al. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics 2013; 194(4), 1029–35. CrossRef
45.Bassett AR, Tibbit C, Ponting CP, Liu JL. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Reports 2013; 4(1), 220–8. CrossRef
46.Yu Z, Ren M, Wang Z et al. Highly efficient genome modifications mediated by CRISPR/Cas9 in Drosophila. Genetics 2013; 195(1), 289–91. CrossRef
47.Senis E, Fatouros C, Grosse S et al. CRISPR/Cas9-mediated genome engineering: an adeno-associated viral (AAV) vector toolbox. Biotechnol. J. 2014; 9(11), 1402–12. CrossRef
48.Mefferd AL, Kornepati AV, Bogerd HP, Kennedy EM, Cullen BR. Expression of CRISPR/Cas single guide RNAs using small tRNA promoters. RNA. 2015; 21(9), 1683–9. CrossRef
49.Cheng R, Peng J, Yan Y et al. Efficient gene editing in adult mouse livers via adenoviral delivery of CRISPR/Cas9. FEBS Letters 2014; 588(21), 3954–8. CrossRef
50.Chiou SH, Winters IP, Wang J et al. Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9-mediated somatic genome editing. Genes Dev. 2015; 29(14), 1576–85. CrossRef
51.Wang D, Mou H, Li S et al. Adenovirus-Mediated Somatic Genome Editing of Pten by CRISPR/Cas9 in Mouse Liver in Spite of Cas9-Specific Immune Responses. Human Gene Ther. 2015; 26(7), 432–42. CrossRef
52.Kabadi AM, Ousterout DG, Hilton IB, Gersbach CA. Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res. 2014; 42(19), e147. CrossRef
53.Fine EJ, Appleton CM, White DE et al. Trans-spliced Cas9 allows cleavage of HBB and CCR5 genes in human cells using compact expression cassettes. Scientific Reports 2015; 5, 10777. CrossRef
54.Truong DJ, Kuhner K, Kuhn R et al. Development of an intein-mediated split-Cas9 system for gene therapy. Nucleic Acids Res. 2015; 43(13), 6450–8. CrossRef
55.Ablain J, Durand EM, Yang S, Zhou Y, Zon LI. A CRISPR/Cas9 vector system for tissue-specific gene disruption in zebrafish. Developmental Cell 2015; 32(6), 756–64. CrossRef
56.Ramakrishna S, Kwaku Dad AB, Beloor J, Gopalappa R, Lee SK, Kim H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res. 2014; 24(6), 1020–7. CrossRef
57.Hendel A, Bak RO, Clark JT et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nature Biotech. 2015 Sep; 33(9), 985–9. CrossRef
58.Sun W, Ji W, Hall JM et al. Self-Assembled DNA Nanoclews for the Efficient Delivery of CRISPR-Cas9 for Genome Editing. Angewandte Chemie 2015 Aug 27; 54(41), 12029–33.CrossRef
59.Polstein LR, Gersbach CA. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nature Chem. Biol. 2015; 11(3), 198–200.CrossRef
60.Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl Acad. Sci. USA 1984; 81(4), 1189–92.CrossRef
http://dx.doi.org/10.1073/pnas.81.4.1189
61.Yu X, Bao B, Echigoya Y, Yokota T. Dystrophin-deficient large animal models: translational research and exon skipping. Am. J. Translational Res. 2015; 7(8), 1314–31.
62.Li D, Qiu Z, Shao Y et al. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nature Biotech. 2013; 31(8), 681–3.CrossRef
63.Feng W, Dai Y, Mou L, Cooper DK, Shi D, Cai Z. The potential of the combination of CRISPR/Cas9 and pluripotent stem cells to provide human organs from chimaeric pigs. Int. J. Mol. Sci. 2015; 16(3), 6545–56.CrossRef
64.Ni W, Qiao J, Hu S et al. Efficient gene knockout in goats using CRISPR/Cas9 system. PloS one 2014; 9(9), e106718.CrossRef
65.Sato M, Miyoshi K, Nagao Y et al. The combinational use of CRISPR/Cas9-based gene editing and targeted toxin technology enables efficient biallelic knockout of the alpha-1,3-galactosyltransferase gene in porcine embryonic fibroblasts. Xenotransplantation 2014; 21(3), 291–300.CrossRef
66.Tan W, Carlson DF, Lancto CA et al. Efficient nonmeiotic allele introgression in livestock using custom endonucleases. Proc. Natl Acad. Sci. USA 2013 Oct 8; 110(41), 16526–31.CrossRef
67.Whitworth KM, Lee K, Benne JA et al. Use of the CRISPR/Cas9 system to produce genetically engineered pigs from in vitro-derived oocytes and embryos. Biol. Reprod. 2014; 91(3), 78.CrossRef
68.Zhou X, Xin J, Fan N et al. Generation of CRISPR/Cas9-mediated gene-targeted pigs via somatic cell nuclear transfer. Cell. Mol. Life Sci. 2015; 72(6), 1175–84.CrossRef
69.Niu Y, Shen B, Cui Y et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 2014; 156(4), 836–43.CrossRef
70.Chen Y, Zheng Y, Kang Y et al. Functional disruption of the dystrophin gene in rhesus monkey using CRISPR/Cas9. Human Mol. Genetics 2015; 24(13), 3764–74.CrossRef
71.Zou Q, Wang X, Liu Y et al. Generation of gene-target dogs using CRISPR/Cas9 system. J. Mol. Cell Biol. 2015;CrossRef
72.Nakamura K, Fujii W, Tsuboi M et al. Generation of muscular dystrophy model rats with a CRISPR/Cas system. Scientific Reports 2014; 4, 5635.CrossRef
73.Hoen PA, de Meijer EJ, Boer JM et al. Generation and characterization of transgenic mice with the full-length human DMD gene. J. Biological Chem. 2008; 283(9), 5899–907.CrossRef
74.Arechavala-Gomeza V, Graham IR, Popplewell LJ et al. Comparative analysis of antisense oligonucleotide sequences for targeted skipping of exon 51 during dystrophin pre-mRNA splicing in human muscle. Human Gene Ther. 2007; 18(9), 798–810.CrossRef
75.Bremmer-Bout M, Aartsma-Rus A, de Meijer EJ et al. Targeted exon skipping in transgenic hDMD mice, A model for direct preclinical screening of human-specific antisense oligonucleotides. Mol. Therapy 2004; 10(2), 232–40.CrossRef
76.Popplewell LJ, Adkin C, Arechavala-Gomeza V et al. Comparative analysis of antisense oligonucleotide sequences targeting exon 53 of the human DMD gene: Implications for future clinical trials. Neuromusc. Disorders: NMD 2010; 20(2), 102–10.CrossRef
77.Xu L, Park KH, Zhao L et al. CRISPR-mediated genome editing restores dystrophin expression and function in mdx mice. Mol. Therapy 2015 Oct 9.CrossRef
78.Bikard D, Jiang W, Samai P, Hochschild A, Zhang F, Marraffini LA. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013; 41(15), 7429–37.CrossRef
79.Qi LS, Larson MH, Gilbert LA, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013; 152(5), 1173–83.CrossRef
80.Albers J, Danzer C, Rechsteiner M et al. A versatile modular vector system for rapid combinatorial mammalian genetics. J Clin. Invest. 2015; 125(4), 1603–19.CrossRef
81.Kennedy EM, Kornepati AV, Mefferd AL et al. Optimization of a multiplex CRISPR/Cas system for use as an antiviral therapeutic. Methods 2015 Aug 17; 15, 30052-9.CrossRef
82.Cheng AW, Wang H, Yang H et al. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013; 23(10), 1163–71.CrossRef
83.Mali P, Aach J, Stranges PB et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nature Biotech. 2013; 31(9), 833–8.CrossRef
84.Perez-Pinera P, Kocak DD, Vockley CM et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nature Methods 2013; 10(10), 973–6.CrossRef
85.Sampson TR, Saroj SD, Llewellyn AC, Tzeng YL, Weiss DS. A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature 2013; 497(7448), 254–7.CrossRef
86.Hale CR, Zhao P, Olson S et al. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 2009; 139(5), 945–56.CrossRef
87.Cong L, Ran FA, Cox D et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013; 339(6121), 819–23.CrossRef
88.McConnell Smith A, Takeuchi R et al. Generation of a nicking enzyme that stimulates site-specific gene conversion from the I-AniI LAGLIDADG homing endonuclease. Proc. Natl Acad. Sci. USA 2009; 106(13), 5099–104.CrossRef
89.Zetsche B, Gootenberg JS, Abudayyeh OO et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015; 163(3), 759–71.CrossRef
90.Koo T, Lee J, Kim JS. Measuring and Reducing Off-Target Activities of Programmable Nucleases Including CRISPR-Cas9. Molecules Cells 2015; 38(6), 475–81.CrossRef
91.Gao Y & Zhao Y. Self-processing of ribozyme-flanked RNAs into guide RNAs in vitro and in vivo for CRISPR-mediated genome editing. J. Integrative Plant Biol. 2014; 56(4), 343–9.CrossRef
92.Athanasopoulos T, Graham IR, Foster H, Dickson G. Recombinant adeno-associated viral (rAAV) vectors as therapeutic tools for Duchenne muscular dystrophy (DMD). Gene Therapy 2004; 11(Suppl. 1), S109–21.CrossRef
93.Nihongaki Y, Yamamoto S, Kawano F, Suzuki H, Sato M. CRISPR-Cas9-based photoactivatable transcription system. Chemistry Biol. 2015; 22(2), 169–74.CrossRef
Affiliations
Marc Moore*, Denis Vallese*, George Dickson & Linda Popplewell§
School of Biological Sciences,
Royal Holloway University of London, Egham, Surrey, TW20 0EX, UK
§Author for Correspondence
linda.popplewell@rhul.ac.uk
*Contributed equally to the work.