
Regulatory viewpoints on the development of advanced stem cell–based medicinal products in light of the first EU-approved stem cell product
10.18609/cgti.2015.010
REGULATORY INSIGHT
Recent years have seen a dramatic increase in research activity and investment into therapeutic applications of stem-cell-based medicinal products. In April 2015, the first stem cell product, ex vivo autologous corneal epithelial cells including stem cells (Holoclar®, Holostem Advanced Therapies), was granted a Marketing Authorization in the European Union (EU). This product is based on limbal stem cells and is intended for the treatment of limbal stem cell deficiency caused by ocular burns. Whilst only one stem cell product has been approved, there are many others being studied for various indications. Approximately 166 clinical trials are testing the use of adult stem cells in regeneration of lost or damaged tissue and in hematological or solid-organ malignancies in the EU. Embryonic stem cells and induced pluripotent stem cells are mainly being explored in nonclinical studies, although early-stage clinical studies with these pluripotent cells have also been reported. Unfortunately, there are also reports of patients travelling outside the EU for unregulated treatments with stem cells. In order to meet patient’s demands for authorized, safe and effective treatments and to foster innovation in this dynamic area, in 2011 the Committee for Advanced Therapies of the European Medicines Agency developed and published regulatory guidance with contributions from European and international experts. It has become clear that a multidisciplinary point of view has to be applied to stem-cell-based products in order to regulate them adequately, thus spanning the bridge from quality criteria and impact of starting materials, animal models, biodistribution and niche, to safety issues including tumorigenicity, and clinical aspects. In this article, we report on the regulatory and scientific framework for stem-cell-based products in Europe and how this can be successfully applied in the development and evaluation of novel stem-cell-based products.
Large expectations have been put on stem cells to provide cures for a multitude of different conditions, including degenerative, inflammatory and metabolic diseases and cancer, as well as the repair and regeneration of damaged or lost tissue. The first stem-cell-based medicinal product in the European Union (EU) was granted a Marketing Authorization in April 2015 for the treatment of limbal stem cell deficiency (LSCD) caused by ocular burns [1]. In cases where treatment options are limited or where no treatment is available, such as degenerative diseases or spinal cord injury, stem cells may provide a promising treatment option. Stem cells are particularly attractive for such diseases as they can provide a renewable and limitless source of cells and production of virtually all cell types. On the other hand, stem cell treatments have been criticized in the scientific literature with respect to safety [2,3] and when used “outside” common scientific principles [4].
In Europe, stem-cell-based therapies are embedded into the legal framework of Advanced Therapy Medicinal Products (ATMPs) [5] and are assessed by the Committee for Advanced Therapies (CAT) at the European Medicines Agency (EMA). Regulation 1394/2007 [5] defines somatic cell therapy, gene therapy and tissue-engineered products as ATMPs. Cell therapy and tissue-engineered products consist of viable cells or tissues, which have either been substantially manipulated or engineered as defined within this legislation (see Art. 2.1(c) of the regulation) [5], or applied in a non-homologous manner (i.e. used for not the same essential function in the recipient and the donor). While developers for ATMPs are expected to fulfil the same regulatory principles as for other medicinal products, specific, tailored requirements for ATMPs have been developed to account for their technical specificities. A reflection paper for stem-cell-based products was developed and published in 2011 [6], based on discussions with relevant working parties and the global scientific and regulatory community, clinicians, patients and physicians. This initial guidance in conjunction with the more general guideline on cell-based medicinal products [7] and the concept of the risk-based approach applied to ATMPs [8] provide a flexible framework for the development of medicinal products using stem cells.
In this article, we discuss the diversity of different stem cells, experiences of their clinical use, the specific regulatory framework for stem-cell-based therapies in the EU and how to best use it for prospective, successful product development. Although Holoclar® is used here as an example and data from the public assessment report is presented, it should be noted that extrapolation to other products has limitations due to differences between products and diseases to be treated.
Diversity of stem-cell-based products
Stem-cell-based products represent a wide spectrum of stem cell-derived cells with variable degrees of self-renewal and differentiation potential, as well as levels of manipulation, availability of scientific knowledge and clinical experience. They include blastocyst-derived embryonic stem cells (hESCs), and adult or somatic stem cells, such as hematopoietic progenitor/stem cells (HSCs), mesenchymal/stromal stem cells (MSCs), and tissue-specific progenitor cells. Cells with stem cell-like characteristics have also been generated from adult differentiated cells by reprogramming to re-acquire self-renewal and differentiation capacity. These include induced pluripotent stem cells (iPSCs) and their intermediate stages [9].
The same features that make stem cells so attractive for therapeutic purposes may also contribute towards their risks. Cells with a high capacity for self-renewal (hESCs and iPSCs) may utilize pathways that facilitate development of tumors or teratomas making them intrinsically tumorigenic (e.g. p53 suppression) [10]. Stem cells also exhibit a variable degree of differentiation capacity. Pluripotent hESCs and iPSCs, for instance, are capable of differentiating along multiple lineages producing all cell types including tissue-specific stem cells, which in turn are mainly responsible for tissue turnover [11]. Stem cell differentiation is also affected by the microenvironment [12]. Typically, HSCs and MSCs have a propensity to home to distant locations by responding to environmental cues [13,14]. Therefore, unintended differentiation or localization to ectopic locations may have serious clinical consequences [15–17].
Extensive clinical experience with unmanipulated bone marrow and peripheral blood-derived stem/progenitor cells already exists. This clinical experience has proven these cell-based therapies to be without serious safety concerns and well tolerated, and the potential risks related to their use are relatively well understood [18]. On the other hand, the clinical experience with pluripotent stem cells is still scarce. The first clinical trial with hESCs was authorized by the FDA in 2009 for patients with spinal cord injury (Clinicaltrials.gov). Since then additional clinical trials with hESC-derived cells have been authorized in the EU and in the USA to treat patients with dry age-related macular degeneration and Stargardt’s macular dystrophy [19]. The limited experience thus far demonstrates that the clinical use of hESC-derived stem cell products does not pose a serious safety risk. In September 2014, the first patient received an iPSC product for the treatment of age-related macular degeneration in Japan [20] and both safety and efficacy results of this trial are eagerly awaited by iPSC developers and regulators.
Stem cells require a tailored regulatory framework
The regulatory framework governing the safe and effective use of stem-cell-based products has to accommodate the heterogeneity in the characteristics, manufacturing and clinical experience of various stem-cell-based products while ensuring that the therapeutic potential and safety concerns typical for this class of products are adequately addressed.
Stem-cell-based products are a special class of cell-based ATMPs, which are considered complex biological medicinal products. One typical feature of these cell-based products is that the medicinal product is defined by the (heterogeneous) starting materials used to manufacture the product. This is due to the fact that characterization and testing of the starting materials, active substance and final product cannot be achieved to the same extent as is usually possible for other medicinal products (e.g. chemical entities). Furthermore, it may be difficult to control the manufacturing process of cell-based products in order to ensure that the product is of consistent quality that can be routinely produced. For advanced stem-cell-based products, the manufacturing process frequently consists of expansion culture steps, which may alter the characteristics and/or functionality of the stem-cell-based medicinal product and which in turn drives the design of the safety and efficacy assessment of clinical studies. Cells may even get phenotypically/genotypically adapted at the end of extensive culture as compared to cells that are in their physiological environment and function. It is therefore important to control the manufacturing process as vigorously as possible and to understand which parameters (e.g., media components, growth factors, culture conditions) may have an impact on the quality and clinical performance of the target cell population [21].
From a clinical-regulatory perspective, the level of manipulation (kind and number of manufacturing steps etc.) is one of the central factors for estimation of risk, which should be addressed in the product development program. In order to manage the level of risk, which may result from the starting material and the manufacturing process, the finished product should nevertheless be controlled and characterized as well as possible with respect to identity, potency, purity and genomic/ phenotypic stability [21]. In the case of stem-cell-based products undergoing genetic modification (be it via integrating or non-integrating vectors or plasmids), this adds further complexity to the manufacturing process and to the product adding uncertainties and hence further needs for characterization and control [22].
As risks, uncertainties and clinical experience associated with different stem cell types may vary significantly, a risk-based approach should be employed during the development of a stem-cell-based therapy [8].
In light of the intrinsic safety concerns surrounding stem cell products, such as tumorigenicity, as well as the limited clinical experience to date, non-clinical safety evaluation may need to be more extensive for stem-cell-based therapies than for conventional biologics or cell-based therapies derived from differentiated, primary cells (e.g., chondrocytes). However, the availability of animal models may be limited due to uncertainty of the similarity between animal and human stem cells or factors involved in the differentiation process. Nevertheless, all efforts should be employed to obtain non-clinical data on the proof-of-concept, biodistribution, and safety in vivo as this information will help to proceed into first-in-man clinical studies. In cases where stem cells have been extensively manipulated ex vivo or nonclinical testing is solely based on homologous animal models, specific safety end points should be included to the first-in-man trial capable of detecting any early toxicity arising from the stem-cell-based product [6]. The degree of caution to be employed when moving into clinical application of stem-cell-based products depends on the estimated risk profile (i.e., tumorigenicity aspects of mesenchymal stromal cells) [23], mode of administration (e.g., local versus systemic), expected biodistribution (e.g., CNS, myocardium), stability of the desired (expected) phenotype and duration of treatment.
Similar to conventional medicinal products, clinical trials should be designed to demonstrate safety and efficacy as well as provide evidence on the proposed mode of action [7,24]. In the design of clinical studies, the disease/indication must also to be taken into consideration and thus the relevant available guidance for clinical studies in the target indication should be followed [25].
Marketing Authorization of Holoclar®: the first approved stem-cell-based
medicinal product in EU
The EMA recommended Holoclar®, the first ATMP containing stem cells, for conditional approval in the EU and the European Commission granted a marketing authorization in February 2015 [1]. Holoclar is used for the treatment of moderate-to-severe LSCD due to physical or chemical burns to the eye(s) in adults. It is the first advanced therapy product recommended for LSCD, a rare eye condition including symptoms of pain, photophobia, inflammation, corneal neovascularisation, loss of corneal transparency, and eventually blindness. This stem-cell based medicinal product is used in the eye to replace damaged corneal epithelium and creating a new reservoir of limbal stem cells (LSCs) for the regeneration of the epithelium. As the number of patients with LSCD due to ocular burns is about 3.3 out of 100,000 people in the EU, the disease is considered ‘rare’ and Holoclar® was designated an ‘orphan medicine’ in November 2008 [1].
A conditional approval is reserved for medicinal products intended for the use in seriously debilitating, life-threatening or rare diseases or in emergency situations in response to public health threats. It means that additional data are required post-marketing in order to generate a comprehensive data set with a view to confirming that the benefit-risk balance is positive.
Holoclar® consists of a transparent circular sheet of viable autologous human corneal epithelial cells, expanded in cell culture that includes LSCs in addition to stem cell-derived transient amplifying and terminally differentiated cells. To manufacture Holoclar® LSCs are derived from a biopsy taken from a small area of undamaged limbus of the patient’s eye. The cells are grown in cell culture with the help of extensively tested irradiated mouse 3T3-J2 feeder cells. The resulting cell suspension is cryopreserved until transplantation is scheduled and thawed cells are used for the manufacture of the final product. To this end, the expanded cells are again seeded on irradiated feeder cells, which have been previously grown on a fibrin support layer. When the cells have expanded and built a confluent layer, the autologous tissue-engineered product is formulated, shipped and administered to the patients.
The marketing authorization holder (MAH) of Holoclar® applied a multidisciplinary approach to the product development, linking the product test results with the clinical data, where possible, and applying the risk-based approach to critical aspects of manufacture and control (i.e., adventitious agents safety testing, stability and transport). The active substance consists of a mixture of cells with an average of 3.5% of LSCs as the main functional component. LSCs were histochemically quantified by expression of the phenotypic marker p63-bright. In addition clonogenic transiently amplifying cells and terminally differentiated corneal epithelial cells are present in the final product [1].
The variability of the cell composition and the amount of functional p63-bright-positive LSCs presented some challenges during product development and assessment. However, the MAH of Holoclar® was justified on the basis of clinical data indicating the minimum amount of LSCs needed for clinical success, as well as the supportive function of the more differentiated cell populations in the formation of the epithelial sheet-like structure needed for the therapy. Cell populations with non-stem cell phenotypes were therefore not regarded as cellular impurities. As the starting material, which is derived from the patient’s own limbus biopsy, is of high value and limited quantity, the characterization strategy needed to be specifically adapted. The colony-forming assay (CFA) was established as a predictor of the clonal/proliferative capacity of the cells, and was used as a sole in-process control (IPC) to control the manufacturing process. Additionally, beside CFA, growth rates are controlled by microscope during cell culture.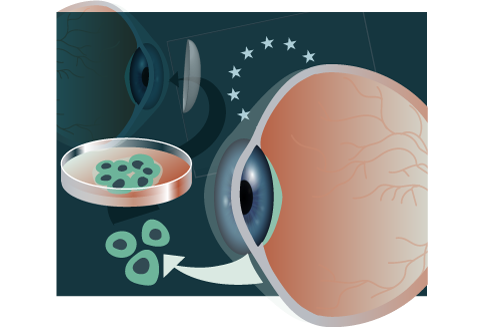
For validation purposes, the MAH of Holoclar® used cadaveric corneas that underwent the same manufacturing process. With the help of this material, important manufacturing aspects such as in-process storage times, and also stability and transport conditions could be defined. Likewise, small manufacturing changes (e.g. qualification of new cell bank, adjustment of specifications) were validated using cadaveric corneas. The finished product is prepared as a transparent sheet of 300,000 to 1,200,000 viable autologous human corneal epithelial cells (79,000–316,000 cells/cm2) attached on a 2.2cm diameter fibrin support in physiological transport medium. Supported by clinical data, the MAH was able to show that the physical organization of the cells on the fibrin sheet is self-assured and different cell components will organize themselves once correctly grafted in vivo. The product is sensitive to mechanical and temperature stress, therefore special emphasis was placed on the packaging and storage conditions. As such, a robust container closure system, consisting of multilayer packaging has been developed for protection of Holoclar®. Transport conditions are tightly controlled and validated beyond current standard pharmaceutical transport controls (e.g., temperature-controlled vehicles with alarm-mode and GPS-tracked remote temperature alarm systems). The shelf life of 36 hours was established based on viability and potency data obtained in stability studies.
The non-clinical development programme of Holoclar® includes supportive information of published studies from scientific literature [1]. Due to the lack of suitable animal models, additional conventional non-clinical studies with Holoclar® were not considered necessary or appropriate and the general concept of transferring LSCs was demonstrated via current transplantation techniques in LSCD. In addition, the shortened development programme was justified by the experience gained from clinical practice with Holoclar® since 1998 [1]. Biodistribution of Holoclar® was addressed by analysis of histological sections of human cornea obtained from patients who had perforating keratoplasty up to 3 years following LSC transplantation. Holoclar® was shown to be effective in restoring a stable corneal surface in patients with moderate or severe LSCD caused by burns in a retrospective study [1]. One year after Holoclar® implantation, 72% of the patients studied (75 out of 104 patients) demonstrate successful implants based on the presence of a stable corneal surface with no or only trace surface defects and little or no ingrown blood vessels. There were reductions in patients’ symptoms, such as pain and inflammation, and also improvements in vision. Despite the absence of a control group, the study outcome was convincing as LSCD does not improve spontaneously.
Key aspects for stem-cell-based medicinal product development
Quality criteria & starting material
Quality requirements for stem cells are particularly difficult to establish due to intrinsic heterogeneity of a cell preparation and also due to the presence of cells at various induced differentiation stages. The quality control of a product is important for both patient safety and efficacy of the product, i.e., viral safety, characterization of cell populations and of the differentiation stage, potency testing and process validation to control for consistency and potential risk of tumorigenicity.
For all cell/tissue starting materials, proper donor testing is required [26]. Thus, viral safety concerns are primarily relevant in relation to “older” ESC lines, where limited information is available on the donors and on the reagents used in cell line derivation and culture. Such reagents, if for example from animal origin, may be the source of inadvertent viral contaminations. For hESCs, a difficult question is what should be the “starting material” for such medicinal products – the blastocysts generated or the embryonic stem cell line developed from them. In general, it is considered that the embryonic cell line should be the starting material and thorough testing may have to compensate for the absence of information in order to address the viral safety concerns. However, testing cannot alleviate the requirements for donor testing on recently developed lines that must comply with the Tissue and Cells legislation [26]. An interesting practical issue pertinent to numerous products in clinical development is the use of mouse feeder cells and the possibility of retroviral transmission by such xenogeneic cells. In case such feeder cells are required and cannot be replaced by a non-xenogeneic alternative, a risk assessment may become the basis for a decision. For Holoclar® an irradiated 3T3-J2 murine feeder cell line is used during the manufacturing process and also as part of the product, which was accepted after the Applicant was able to confirm the absence of proliferation of the irradiated cells and lack of any safety signals related to these feeder cells. Murine 3T3-J2 clones have been used for example in the treatment of patients with epithelial defects, such as full-thickness burns [27]. Recently, fully tested master cell banks of these feeder cells have become available from various manufacturers (e.g., Waisman biomanufacturing).
The stem cell preparations often contain mixed populations, which may be further impacted by the in vitro cell culture process. Therefore, specific markers and/or cell characteristics need to be identified in order to be able to follow for example, differentiation/redifferentiation processes of the stem cells. It has been shown that under appropriate culture condition keratinocytes can generate holoclones, paraclones and meroclones that are all essential components of the product [28]. From these holoclones arise the stem cells that are essential to permanently restore epithelial defects. Holoclones have also been located in the limbus and are identifiable by the expression of the p63 transcription factor [29]. For Holoclar®, it was demonstrated that a specified amount of stem cells (p63-bright cells) must be present in the final product for clinical effect and this marker was further used as a surrogate marker for potency.
Although there is a lot of information about the characteristics of certain stem cells, it is still unclear whether epigenetic markers could play a role in defining “safe” stemness and lineage commitment of stem cells. It is also not clear how well different stem cells maintain their differentiated phenotype after in vitro differentiation and whether these could revert back to undifferentiated or even pluripotent stage in in vivo conditions.
The possibility of separating undesirable cells, as well as the potential to include purification steps depends on the starting material and the manufacturing process. Whilst for undifferentiated hESCs there are a several relevant markers that allow predictive assessment of the number of pluripotent cells entering differentiation, it may be more difficult to define a meaningful marker pattern after differentiation. Expression of various markers of cell fate might be on or off at different time points or, in the early stages of differentiation, co-expressed with markers of pluripotency (proliferation). Kinetic studies of relevant markers could be a possible approach to validate the process of lineage commitment. For the comparison of lineage-committed and fully differentiated cells functional assays could also be valuable tools.
Regarding purity, an appropriate balance needs to be determined between heterogeneity, where necessary, and consistency of the product required to achieve the desired therapeutic effect [21]. The use of selective positive markers for target cells and negative markers for contaminant cells may be one approach. While on a research scale such approaches (e.g. by fluorescence-activated cell sorting) may be possible, their scalability to a level required of a medicinal product may be challenging. On the other hand, the Holoclar® case has convincingly demonstrated that a cell population consisting of differentiated cells and stem cells can deliver the functionality, in concert, whereby the differentiated cells contribute to the forming of the epithelial-like structure harboring the stem cells. Thus, there was no need to identify and control certain non-stem cells as cellular impurities, although the ratio of stem/non-stem cells is important for other purposes (potency and process consistency). For tissue-engineered products like Holoclar® it is important to note that all IPC and release testing will require destructive sampling, possibly impacting the final product properties. This should be considered and novel, non-invasive strategies (e.g., based on factors secreted by the cells in culture medium) may be more suitable to be developed for such products.
One of the most discussed issues for all cell-based products is the availability of a good, relevant candidate potency test. It is generally accepted that a candidate potency assay should already be available at the early stages of product development and preferably validated before entering pivotal clinical trials, since clinical performance may then be better captured and correlated with the potency test [7]. Potency should reflect the mode of action of the product, although it is acknowledged that this might not always be possible. For example, the mechanism of action of bone marrow cells for cardiac repair might be hard to define and not yet known, based on current scientific knowledge [30]. Therefore, multiple approaches drawn from biological and functional characterization of the cells may, on a case-by-case basis, offer a solution for adequate testing of potency. Assays based on colony-forming efficacy may prove useful as predictors of clonogenic and proliferative capacity as one feature of a product based on heterogeneous cell population, as seen with Holoclar®.
Given the inherent risk of tumorigenicity related to stemness versus lineage commitment, the ability to assess tumorigenicity at the level of process development should be utilized. In general, karyotypic stability, genetic or epigenetic instabilities or transcriptional changes are best addressed at a nonclinical level; however, if recurrent genetic abnormalities are identified in the studies, further characterization and release testing as part of quality control may be necessary.
Where commercial manufacturing of stem-cell-based products is intended, one should be prepared for possible manufacturing changes, which in turn may impact the product characteristics and/or functionality. To address the impact of such changes, comparability of the product pre- and post-change should be evaluated. This may be particularly challenging for stem-cell-based products with multiple characteristics to be followed. This further highlights the need for thorough characterization of the product and identification of the key quality attributes that are imperative for the intended use of the product [21].
Non-clinical aspects
Non-clinical development of stem-cell-based medicinal products is the same, in principle, as for any other medicinal product whereby it is assessed whether the applicant has provided credible and satisfactory answers to two questions:
1. What evidence is presented that supports the expectation of therapeutic benefit (proof-of-principle, pharmacodynamic and pharmacokinetic properties)?
2. What evidence is presented that supports the expectation of safety (e.g. immunogenicity, tumorigenicity)? [6,7]
The design of non-clinical studies should be driven by a science-based approach (rather than uncritically testing the product) involving available knowledge and assessment of risk, including various factors: type and available knowledge of the product, its degree of manipulation, the availability of a relevant (homologous) animal model (relevant both as regards resemblance of the medicinal product in question and the results that are to be expected) and type of administration (e.g., part of surgical procedure or by a catheter).
Due to the nature of these products, they respond to the environment in which they are administered and the non-clinical testing must take this into account, as well as the differences between species [6]. The level of manipulation of the cells also dictates some of the requirements for the non-clinical testing and both in vitro and in vivo approaches should be considered.
Animal models
For stem-cell-based products it is likely that more than one animal model may be necessary to cover all safety aspects, and both the human cells and homologous animal cells may need to be used. The choice of the most relevant animal model should be determined by the specific aspect to be addressed in the study. However, even the best animal model is only a model. Predictivity of animal data and meaningful estimation of risk necessitates careful interpretation of data and thorough understanding of the values and limitations of each model. Non-clinical studies in non-relevant species may be misleading and are discouraged from a scientific point of view and in view of animal welfare.
The choice of the animal model(s) used for development of stem-cell-based therapies and in particular to study proof-of-principle and safety, may be complicated due to species differences. For all animal studies it is important to determine which information can actually be extracted from studies in animals, and which animal model (homologous or heterologous) is most suited to obtain this information. In homologous animal models, animal cells are used in the animal model to “simulate” the human cell-based medicinal product, as opposed to heterologous models where the human stem cell product is applied in the animal model. In the latter approach, the actual medicinal product is tested, not taking the species differences into account that may limit the relevance of the obtained results.
In addition to considerations of the relevance of such models, the requirement of designing a homologous animal model depends on the nature of the stem-cell-based product itself: if there is no human experience with the product or similar products so far, then there may be a higher risk attributed to such a product, and then the use of a relevant homologous model, if available, for proof-of-principle studies would be valuable to study the potential effects and problems that may occur later in the clinic. Even though it is recommended that safety data are included in studies with homologous animals, some specific data, such as biodistribution and ectopic localization may better be obtained with the actual human product itself, since its behavior may differ from the animal analogue.
While for some (somatic or multipotent) stem cell-derived products important information can be obtained from homologous animal models, for embryonic (pluripotent) stem-cell derived products the relevance of a homologous mouse model may be limited given the known differences between human and mouse ESCs [31]. For such products, studies in a heterologous setting using immunocompromised animals and/or alternative in vitro studies using human tissue systems/cells may be more suitable. To establish the relevance of an animal model, the (patho)physiological characteristics of the animal should also be taken into consideration; this may warrant the use of a large animal model that better resembles the human situation. Large animals may also be needed when the route of administration, e.g., surgical procedure, is not feasible in a small animal model.
In the case of Holoclar®, conventional non-clinical animal studies have not been considered appropriate or feasible due to the unique and diverse structural and biological properties of the product and therefore an abridged non-clinical programme was accepted [1]. The proof-of-principle was demonstrated in non-clinical studies in rabbits, which showed that ex vivo-expanded limbal stem cells cultured on a fibrin support can be used to replace and regenerate lost corneal epithelium [32,33]. The cells were able to create a structural replacement, with the formation of a normal thickness corneal epithelial cell layer. The functionality of these types of grafts was further supported by the demonstration of clinical efficacy, as defined by a resolution of LSCD-associated symptoms (ocular burning and pain, photophobia, foreign body sensation) as well as improved visual acuity [34]. For the pharmacokinetic data, the Applicant referred to published data [35], in which the distribution of a similar cell sheet based on skin keratinocytes was analyzed after subcutaneous transplantation in athymic mice.
Similarly, non-clinical toxicology assessment of Holoclar® was limited and abridged due to the lack of relevant animal models with similar ocular structure, and focussed on the characterization of the tumorigenic and carcinogenic potential of the therapy. Other aspects of toxicology, such as antigenicity and microchimerism due to the presence of the irradiated 3T3-J2 feeder layer, were largely deduced from clinical findings [1].
Biodistribution & niche
Successful therapeutic outcome, but also the safety profile, of a stem-cell-based medicinal product administered to humans depends on the extent of biodistribution into the whole organism and target tissue from the site of administration (organ or tissue). Stem cells that reach the target tissue adapt to the new microenvironment (niche) and express their biological activity effectively regardless of their intended use, be it pharmacological or repair, restoration or regeneration of a tissue or function. Therefore, whole body distribution patterns and microenvironmental influencing of stem cell fate are considered critical factors. Unfortunately, in most cases, it is still not known which factors drive and influence biodistribution and homing of stem cells in a given tissue. Various factors including the target tissue (e.g., bone marrow, liver or heart), the route of stem cell administration, stem cell load, physical contact/adhesion with other cell types or macromolecules and chemo-attractants influence the fate of the administered cells [36]. Rolling and firm adhesion of stem cells to the endothelial cells as well as transendothelial migration across physical barriers are important determinants for biodistribution to target tissue [37].
Stem cells can then remain as undifferentiated, self-renew, or give rise to differentiated progeny needed for the intended function. To estimate efficacy and safety of stem-cell-based medicinal products, the biodistribution pattern and further fate of the cells should ideally be evaluated. Furthermore, there is a risk of ectopic tissue formation and unwanted differentiation in cases where the administered cells respond to the environmental cues in an unwanted manner, as for example was observed with the development of severe calcification in patients with acute myocardial infarction after administration of bone marrow cells [38]. In animal models, quantitative polymerase chain reaction (qPCR) techniques can be used to detect administered cells in resected tissues and organs. In xenotransplantation, human cells can be detected in animals using human-specific sequences [39]. Non-invasive imaging technologies could be used to track stem cell migration patterns in vivo (in relevant animal models and/or in humans) as the cells move from the site of administration or implantation through the whole body towards the tissue of action. Over the past years, various non-invasive bioimaging techniques such as multiphoton intravital miscoscopy (MP-IVM), nuclear magnetic resonance (NMR), magnetic resonance imaging (MRI), combined single-photon emission CT (SPECT/CT) scanning microscopy, and the use of radionuclide (111In, 99Tc) have been developed for quantitative tracking of stem cell transplants [40]. Moreover, immunofluorescence/bioluminescence resulting from reporter genes transferred into stem cells and expressed fluorescent proteins such as GFP, firefly luciferase can also be utilized. State-of-the-art techniques should be used based on the expected amount of information and the perceived level of risk of a particular product. Likewise, some techniques including reporter genes may only be feasible in a non-clinical model. If such an approach is chosen, special attention should be paid to the comparability of the labelled product with the unlabelled product. Developers of stem-cell-based medicinal products may have to focus their biodistribution studies on relevant animal models even though the results can be applied to humans by extrapolation only. An unresolved question is how to effectively monitor long-term behavior of cells in humans.
Tumorigenicity
A common safety concern related to stem cell therapy is the occurrence of tumorigenicity. Tumor development may be linked to product-related factors such as cell source, extent of ex vivo manipulation, use of excipients, and/or to indication or patient related-factors including the implantation site. The formation of teratomas, an intrinsic characteristic of hESC and iPSC when injected in vivo, may be linked to the lack of suitable microenvironment cues rather than to transformation as the same cells differentiate normally into all type of tissues when injected into blastocysts [41]. The capacity of stem cells to give rise to uncontrolled growth or precancerous tissue may be affected by the cell status, the implantation site and the aggregation state of these cells as well as potentially acquired genomic instability during in vitro expansion and differentiation. How to differentiate between senescence and malignant transformation is still scientifically debated [42]. It might be impossible to separate tumorigenic cells from non-tumorigenic cells, as pluripotency and tumorigenicity appear to be interconnected, i.e., activation of tumor suppressor gene p53 in ESCs leads to their rapid differentiation [43] and down regulation of p53 seems to be associated with increased iPSC generation [10].
To address these safety concerns, further studies to analyze the tumorigenic potential are required and both in vitro and in vivo studies should be performed in order to obtain information on the risk level. General features of the cell population such as growth factor dependency and regulatory signalling pathways may be studied by molecular and biochemical technologies. Cytogenetic techniques such as fluorescence in-situ hybridization (FISH) or subtelomere screening may be useful, and additional novel molecular techniques are emerging. In the case of Holoclar® karyotype analysis, soft agar assays and growth factor dependence growths were examined [1]. These tests suggested a low tumorigenic potential of Holoclar®, which was supported by clinical data, but the number of patients in the studies was too low to fully exclude this risk. Therefore, further monitoring of the safety of Holoclar® in clinical practice is required as part of the risk management of Holoclar®, including post-marketing reports of neoplasms occurring after implantation as well as long-term safety data from a registry and a clinical trial.
Animal models can also be used for the development of biological functional assays and for long term toxicity studies. The extent of testing is determined by available techniques, the nature of the product (e.g., autologous vs allogeneic), and the amount of cells available. It is also important to develop data-based acceptance criteria and not only to rely on descriptive findings. It has also been highlighted that protocols for cell differentiation should be species specific. Therefore, ideally, human stem-cell-based medicinal products should be tested for tumorigenicity in a suitable immunocompromised animal model. Extrapolation from animal tumorigenicity data to humans is demanding, and final proof can only be obtained from clinical use. The clinical trial protocol should include careful dosing, monitoring and suspension rules. Interpretation of these results is particularly challenging and may indicate an inherent risk of tumorigenicity. To manage uncertainty that remains after administration to patients, rescue strategies to remove or kill the administered cells in the patient (such as inclusion of a suicide gene) could serve as an alternative to an extended tumorigenic analysis.
Clinical aspects
The clinical development of stem-cell-based products is complicated by a number of issues, since established principles such as pharmacodynamic, pharmacokinetic and dose finding may have to be adapted to the specific nature and needs of the product in question.
In early clinical development, lack of non-clinical safety studies to assess the risk imposed on the enrolled subjects may pose significant challenges. Although fewer risks may be perceived for autologous versus allogeneic cell sourcing, manipulation of autologous cells may increase the risks even beyond what may be foreseen for unmanipulated allogeneic cells. Furthermore, the risks depend greatly on the mode of administration and the clinical condition. It is unclear to what extent the immune system tolerates manipulated autologous cells, and is able to control, for example, excessive in vivo growth of the cells administered. Consequently, it may be challenging to define the first-in-human starting dose of acceptable tolerability and safety. The most common adverse reactions with Holoclar® are eye disorders, including blepharitis and corneal epithelium defect [1]. Adverse reactions related to the surgical procedure and required concomitant corticosteroid regimen include conjunctival haemorrhage and glaucoma, respectively.
Furthermore, the relationship between efficacy and dose administered should be understood. From a trial subject’s perspective this is particularly important as patients hope to obtain a benefit from the treatment. The feasibility of dose-finding studies, however, is dependent on suitable clinical read-outs sufficiently sensitive to detect clinical differences in efficacy between different doses. It might be possible that living cells, applied as a medicinal product, may have sufficient plasticity to divide and fill out the defect, even if the dose initially applied was too small. However, such considerations should be supported by relevant clinical data. Absence of dose-finding studies may be problematic if the pivotal clinical study only demonstrates borderline results or significant safety findings that may potentially have been avoidable with a lower, albeit still efficacious dose. This concern is one of the key points when developing a completely new medicinal product. For Holoclar® dose-finding studies were not formally carried out. The authorized dose was supported by the results of the three retrospective, multicenter, case series-based, non-randomized, and uncontrolled observational studies [1]. Furthermore, studies carried out by Pellegrini and colleagues [44,45] (in two patients [grafts contained about 2.0×106 cells]) provided insights into how the sourcing, procurement and testing, as well as the dose, were optimised in the early stages of the clinical development program. For Tissue Engineering Products (TEP) like Holoclar® the meaning of “dose” may be difficult to define. For many TEP applications the amount of the product administered is influenced by factors such as trauma, extent or size, and the intended effect like regeneration, replacing or restoring of target functions or tissue with the time to steady-state as a dependent variable. Thus it is essential that applicants detail the description of the measures taken to avoid/minimize systematic errors in the study design, conduct, analysis, and interpretation of the results submitted. The product consists of a confluent sheet of 300,000–1,200,000 viable autologous human corneal epithelial cells (79,000–316,000 cells/cm2), including on average 3,5% (0.4–10%) LSCs. The specifications for cell density and potency are controlled during the manufacturing process of the product. The same specifications were already applied for the treatment of patients included in the retrospective studies which demonstrated successful ocular surface reconstruction and replacement of LSCs and related barrier function 1 year after Holoclar® implantation. In the pivotal trial, 72% (75/104) of patients were considered a treatment success with no or trace epithelial defects and vessel penetration, although some patients (12 out of 104 patients in the pivotal trial) required more than one graft. Clinically meaningful outcomes also included a reduction in the proportion of patients with symptoms from 39% (40/104) to 12% (12/104) as well as vision gains equivalent to three lines on a vision chart in 39% (40/114) of patients. Furthermore, patients with deep stromal scarring were shown to be more likely to have a successful keratoplasty subsequent to Holoclar® treatment.
Another experience of stem cell products is the Health Canada approval of a MSC therapy intended for the treatment of acute graft-versus-host disease (GvHD) in children who have failed all previous therapies for GvHD following bone marrow transplantation [46]. In the EU, similar products have been classified as somatic cell therapy medicinal products by the CAT [47]. The authorization was provided by Health Canada with conditions, such as expecting further proof of efficacy to be submitted post-authorization.
Stem cell products have created an as yet unresolved debate on the length of follow-up required to determine efficacy, which is not only linked to short-term efficacy and safety but also to the intended final effect, both at cellular and structural level in the recipient’s body (e.g., integration of TEPs). Therefore, the length of follow-up studies should be considered both from a safety and efficacy perspective, taking into consideration all relevant factors. Article 14 of the Advanced Therapies Regulation [5] offers the possibility of post-authorization follow-up of efficacy studies (in addition to safety) as a specific obligation, which enables the approval of ATMPs in the EU that have a positive benefit/risk but where limited information is available on long-term outcome. Defining long-term efficacy may be challenging, since the natural course of the underlying disease may at some point prevail, and gradual loss of efficacy may or may not be problematic as regards re-definition of benefit/risk. For Holoclar®, long-term data up to 10 years, although limited, suggested persistence of the treatment effects over time.
Lastly, applicants should note that within the EU (as well as globally) ethical issues exist within the competencies of Member States and therefore must be taken into account when considering different approaches in hESC and iPSCs. The diversity of legislative provisions among Member States on hESC research must also be taken into consideration; 17 Member States permit – subject to their oversight and conditions – research involving hESCs, 7 are restrictive (Croatia, Germany, Italy, Lithuania, Malta, Romania and Slovakia) [48] and the remainder have no specific legislation. This makes commercialization of hESC-based products particularly challenging.
Conclusions
Stem cell therapies utilise a wide spectrum of stem-cell-based products with variable degrees of self-renewal and differentiation potential, as well as availability of scientific knowledge and clinical experience. Consequently, risks and expected benefits associated with different stem cell products may vary significantly. Stem cells are considered particularly attractive in therapeutic applications where no other treatment options are available (e.g., spinal cord injuries, LSCD). In Europe, the pathway for safe and efficacious development and application of stem-cell based medicines has been mapped with specific guidance and approval of the first stem cell medicinal product in the EU. The development of Holoclar® was based on a multidisciplinary and risk-based approach, which proved to be a successful way to bring such products to the EU market. However, it must be noted that the post-marketing generation of additional efficacy and safety data, including a prospective interventional study is expected as part of the conditional approval.
The challenges relating to the development of stem-cell-based products are multi-faceted and require thorough planning and solid research before entering into the clinical development pathway. The heterogeneity of the starting materials, challenges in manufacturing and quality control, limitations in non-clinical studies and the requirements for the clinical studies should be carefully explored to build a bridge from the quality of the product to successful clinical development. A risk- and knowledge-based approach should always be the basis for decision making.
Tumorigenicity is clearly one of the biggest inherent risks of these products, especially where pluripotent cells are used. This will have to be taken into account during the development of such products, but also in benefit–risk estimations during assessment of clinical trial and marketing authorization applications. The risk level may vary considerably depending on the cell source, level of manipulation, route of administration, and clinical condition including its prognosis and therapeutic alternatives. The more good quality data gathered on the actual tumorigenicity of different stem-cell-based products, the easier it will be to find acceptable criteria for the risk of tumorigenicity of these products.
The multidisciplinary perspective suggests that clinical results will relate not only to “usual” factors such as dose, but also to the status of the surrounding tissue, quality aspects of the cells, method/route of administration, extent and nature of manipulation, clinical condition for example. Long-term follow-up requirements will always depend on the patient population and on the risks perceived, but likewise present an opportunity to more firmly establish benefit for patients. Based on the framework developed and the experience collected it is foreseen that the primary challenges of stem-cell-based medicines are:
1. Heterogeneity of both product and clinical condition
2.Limitations of traditional methodologies, which can only be met with a multidisciplinary, science- and risk-based approach.
Interpretation of existing regulatory principles for the wide variety of possible stem-cell-based products requires a close dialogue between regulators, developers and patients.

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.
Financial and competing interests disclosure
The authors have no relevant financial involvement with an organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock options or ownership, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Disclaimer
The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of the EMA or one of its Committees or Working Parties.
Acknowledgments
The authors wish to thank members of the former Cell-Products Working Party (CPWP), who participated in the preparation of the reflection paper.
Affiliations
Egbert Flory1,16, Paolo Gasparini2,16, Veronika Jekerle3, Tiina Palomäki4,16, Patrick Celis3,16, Tomáš Boráň5,16, James W McBlane6,16, John Joseph Borg7,16, Jan Kyselovic8,16, Metoda Lipnik-Stangelj9,16, Toivo Maimets10,16, Margarida Menezes-Ferreira11,16, Guido Pante12, Stefanie Prilla3, Una Riekstina13,16, Christian K Schneider14,16, Asterios Tsiftsoglou15,16 & Paula Salmikangas4,16.
1Medical Biotechnology Division, Paul-Ehrlich-Institut, Langen, Germany
2University of Trieste, Trieste, Italy
3European Medicines Agency (EMA), London, UK
4Finnish Medicines Agency, Helsinki, Finland
5State Institute for Drug Control, Prague, Czech Republic
6Medicines and Healthcare Products Regulatory Agency (MHRA), London, UK
7Awtorità dwar il-Mediċini, Post-Licensing Directorate, Malta
8Comenius University, Dept. of Pharmacology and Toxicology, Slovakia
9University of Ljubljana, Faculty of Medicine, Ljubljana, Slovenia
10University of Tartu, Tartu, Estonia
11Infarmed – National Authority of Medicines and Health Products, Lisboa, Portugal
12Italian Medicines Agency, Rome, Italy
13University of Latvia, Riga, Latvia
14Danish Health and Medicines Authority, Copenhagen, Denmark
15Aristotle University of Thessaloniki, Thessaloniki, Greece
16Current and former members of the Committee for Advanced Therapies (CAT)
References
1. Holoclar, European Public Assessment Report (2015)
Website
2. Thirabanjasak D, Tantiwongse K, Thorner PS. Angiomyeloproliferative lesions following autologous stem cell therapy J. Am. Soc. Nephrol. 2010; 21: 1218–22.
CrossRef; PMCid
3. Amariglio N. Donor-Derived Brain Tumor Following Neural Stem Cell Transplantation in an Ataxia Telangiectasia Patient. PLoS Med. 2009; 6: e29.
CrossRef; PMCid
4. CAT Committee: Use of unregulated stem-cell based medicinal products. Lancet 2010; 376: 514.
CrossRef
5. Regulation (EC) No 1394/2007 of the European Parliament and of the Council on advanced therapy medicinal products and amending Directive 2001/83/EC and Regulation (EC) No 726/2004. OJ L324, 121–137 (2007).
6. EMA/CAT Reflection paper on stem cell-based medicinal products EMA/CAT/571134/2009.
Website
7. EMEA/CHMP/CPWP Guideline on Human Cell-based Medicinal Products EMEA/CHMP/410869/2006.
Website
8. EMA/CAT/CPWP Guideline on the risk-based approach according to annex I, part IV of Directive 2001/83/EC applied to Advanced therapy medicinal products (EMA/CAT/CPWP/686637/2011)
Website
9. Takahashi K & Yamanaka S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006; 126: 663–7.
CrossRef; PMid
10. Maimets T, Neganova I, Armstrong L and Lako M. Activation of p53 by nutlin leads to rapid differentiation of human embryonic stem cells. Oncogene 2008; 27, 5277–87.
CrossRef; PMid
11. Robinton DA & Daley GQ. The promise of induced pluripotent stem cells in research and therapy. Nature 2012; 481(7381): 295–305.
CrossRef; PMCid
12. Scadden DT. The stem-cell niche as an entity of action. Nature 2006; 441: 1075–79.
CrossRef; PMid
13. Méndez-Ferrer S, Michurina TV, Ferraro F et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010; 466: 829–34.
CrossRef; PMCid
14. Zhang, J, Niu C, Ye L et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003; 425: 836–84.
CrossRef; PMid
15. Breitbach M, Bostani T, Roell W et al. Potential risks of bone marrow cell transplantation into infarcted hearts. Blood 2007; 110(4): 1362–9.
CrossRef; PMid
16. Jeong JO, Han JW, Kim JM et al. Malignant tumor formation after transplantation of short-term cultured bone marrow mesenchymal stem cells in experimental myocardial infarction and diabetic neuropathy. Circ. Res. 2011; 108(11):1340–7.
CrossRef; PMCid
17. Amariglio N(1), Hirshberg A, Scheithauer BW et al. Donor-derived brain tumor following neural stem cell transplantation in an ataxia telangiectasia patient. PLoS Med. 2009; 17;6(2):e1000029. CrossRef
18. Hatzistergos KE, Blum A, Ince T, Grichnik JM, Hare JM. What is the oncologic risk of stem cell treatment for heart disease? Circ. Res. 2011; 108(11):1300–3.
CrossRef; PMCid
19. Schwartz, SD, Regillo, CD, Lam, BL et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: follow-up of two open-label phase 1/2 studies. Lancet 2015; 385 (9967): 509–16.
CrossRef
20. Cyranoski D. Next-generation stem cells cleared for human trial. Nature 2014; doi:10.1038/nature.2014.15897
CrossRef
21. Salmikangas P, Menezes-Ferreira M, Reischl I et al. Manufacturing, characterization and control of cell-based medicinal products: challenging paradigms toward commercial use. Regen Med. 2015; 10(1): 65–78.
CrossRef; PMid
22. EMA/CAT Guideline on quality, non-clinical and clinical aspects of medicinal products containing genetically modified cells EMA/CAT/GTWP/671639/2008.
Website
23. Barkholt L, Flory E, Jekerle V et al. Risk of tumorigenicity in mesenchymal stromal cell-based therapies-bridging scientific observations and regulatory viewpoints. Cytotherapy 2013; 15(7): 753–9.
CrossRef; PMid
24. Commission Directive 2009/120/EC of amending Directive 2001/83/EC of the European Parliament and of the Council on the Community code relating to medicinal products for human use as regards advanced therapy medicinal products. OJ L243; 3–12 (2009).
25. EMA clinical safety and efficacy guidelines.
Website
26. Directive 2004/23/EC of the European Parliament and of the Council on setting standards of quality and safety for the donation, procurement, testing, processing, preservation, storage and distribution of human tissues and cells. OJ L 102, 48–58 (2004).
27. Wright KA, Nadire KB, Busto P, Tubo R, McPherson JM, Wentworth BM. Alternative delivery of keratinocytes using a polyurethane membrane and the implications for its use in the treatment of full-thickness burn injury. Burns 1998; 24(1): 7–17.
CrossRef
28. Pellegrini G, Ranno R, Stracuzzi G et al. The control of epidermal stem cells (holoclones) in the treatment of massive full-thickness burns with autologous keratinocytes cultured on fibrin. Transplantation 1999; 68(6): 868–79.
CrossRef; PMid
29. De Luca M, Pellegrini G, Green H. Regeneration of squamous epithelia from stem cells of cultured grafts. Regen Med. 2006; 1(1): 45–57.
CrossRef; PMid
30. Boyle AJ, Schulman SP, Hare JM. Stem cell therapy for cardiac repair ready for the next step. Circulation 2006; 114: 339–52.
CrossRef; PMid
31. Ginis I, Luo Y, Miura T et al. Differences between human and mouse embryonic stem cells. Dev. Biol. 2004; 269(2): 360–80.
CrossRef; PMid
32. Gimeno LF, Lavigne V, Gatto S, Croxatto JO, Correa L, Gallo JE. Advances in corneal stem- cell transplantation in rabbits with severe ocular alkali burns. J. Cataract Refractive Surge. 2007; 33(11): 1958–6.
CrossRef; PMid
33. Talbot M, Carrier P, Giasson CJ et al. Autologous transplantation of rabbit limbal epithelia cultured on fibrin gels for ocular surface reconstruction. Mol. Vis. 2006; 12: 65–75.
PMid
34. Rama P, Matuska S, Paganoni G, Spinelli A, De Luca M, Pellegrini G. Limbal stem-cell therapy and long-term corneal regeneration. N. Engl. J. Med. 2010; 363(2): 147–55.
CrossRef; PMid
35. Di Nunzio F, Maruggi G, Ferrari S et al. Correction of laminin-5 deficiency in human epidermal stem cells by transcriptionally targeted lentiviral vectors. Mol. Ther. 2008; 16(12): 1977–85.
CrossRef; PMid
36. Gattazzo F, Urciuolo A, Bonaldo P. Extracellular matrix: A dynamic microenvironment for stem cell niche. BBA 2014; 1840(8): 2506–19.
CrossRef
37. Liu L, Eckert MA, Riazifar H, Kang D-K, Agalliu D, Zhao W. From Blood to the Brain: Can Systemically Transplanted Mesenchymal Stem Cells Cross the Blood-Brain Barrier? Stem Cells Int. 2013; 2013: 435093.
38. Yoon,Y-S, Park J-S, Tkebuchava T, Luedeman C and Losordo DW. Unexpected Severe Calcification After Transplantation of Bone Marrow Cells in Acute Myocardial Infarction. Circulation 2004; 109: 3154–7.
CrossRef; PMid
39. Toupet K, Maumus M, Peyrafitte J-A et al. Long-term detection of human adipose derived mesenchymal stem cells after intra-articular injection. Arthritis & Rheumatism 2013; 65(7): 1786–94.
CrossRef; PMid
40. Sensebé L & Fleury-Cappellesso S. Biodistribution of mesenchymal stem/stromal cells in a preclinical setting. Stem Cells Int. 2013; 2013: 678063.
CrossRef; PMCid
41. Blum B & Benvenisty N. The tumorigenicity of diploid and aneuploid human pluripotent stem cells. Cell Cycle 2009; 8 (23): 3822–30.
CrossRef; PMid
42. Prockop DJ. Defining the probability that a cell therapy will produce a malignancy. Mol. Ther. 2010; 18: 1249–50.
CrossRef; PMCid
43. Hong H, Takahashi K, Ichisaka T et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 2009; 460: 1132–5
CrossRef; PMCid
44. Pellegrini G, Traverso CE, Franzi AT, Zingirian M, Cancedda R, De Luca M. Long-term restoration of damaged corneal surfaces with autologous cultivated corneal epithelium. Lancet 1997; 349(9057): 990–3.
CrossRef
45. Pellegrini G, Golisano O, Paterna P et al. Location and clonal analysis of stem cells and their differentiated progeny in the human ocular surface. J. Cell Biol. 1999;145(4): 769–82.
CrossRef
46. Health Canada Report of the Expert Advisory Panel on Prochymal 2012.
Website
47. CAT classification reports
Website
47. Human Stem Cell Research and Regenerative Medicine, focus on European policy and scientific contributions 2013.
Website