Establishment of a scalable manufacturing platform for in-silico-derived ancestral adeno-associated virus vectors
Cell Gene Therapy Insights 2018; 4(S1), 753-769.
10.18609/cgti.2018.080
Adeno-associated virus (AAV) is an effective viral vector for gene therapy due to its non-pathogenicity, high transduction efficiency, broad tissue specificity and long-term transgene expression activity. As such, AAV has become a vector of choice with over 120 active clinical trials worldwide. With this increased demand also comes the need for scalable GMP manufacturing processes that are robust and can support large-scale production. However, current manufacturing of AAV vectors for gene therapy is labor-intensive, resulting in high production costs (adherent-based process) or taking much longer to establish (e.g., producer cell line). Here we report the establishment of a scalable manufacturing platform for AAV production based on transient triple transfection of suspension HEK293 cells. Through flow cytometry, limiting dilution and productivity screening, we first isolated a high producer clonal suspension HEK293 cell line. After medium selection and transfection optimization, we scaled up the production process to 50-Liter using single use bioreactors (SUB). The platform was initially developed for a novel AAV vector (Anc80), that belongs to a large family of novel AncAAV’s, as described previously by Luk Vandenberghe [1] but has also been used for the successful production of other AAV serotypes. Using affinity chromatography followed by ion exchange chromatography, we successfully purified the recombinant AAV/Anc80 products with a high percentage of full viral particles. Taken together, we have established an effective, cost-efficient and scalable manufacturing process for AAV/Anc80 that supports the ever-growing production demands in the AAV gene therapy market.
Submitted for Review: Aug 29 2018 Published: Oct 17 2018
Introduction
AAV has emerged as a key gene therapy vector due to its unique characteristics, including a high safety profile and long-term gene expression activity in humans [2]. There are more than 120 clinical trials using AAV vectors worldwide and the first AAV-based gene therapy in the United States for inherited retinal disease was approved in December of 2017. The fast-growing demand for recombinant AAV in gene therapy highlights the requirement for emerging technologies to develop scalable and improved AAV manufacturing platforms [3].
AAV is a ~25-nm, non-enveloped and non-autonomous virus from the family of Parvoviridae [2]. The AAV genome is composed of 4.7 kb linear single-stranded DNA that comprises genes encoding for replication (Rep), capsid (Cap), and virus assembly (AAP) proteins, flanked by inverted terminal repeats (ITR) on both ends. However, these genes are not sufficient for AAV replication in host cells. The reproduction of AAV is dependent on the co-infection of helper viruses, such as Adenovirus, Herpes Simplex Virus, etc. Specifically, five Adenovirus genes were identified that were necessary and sufficient for AAV production in permissive cells, i.e., E1A, E1B, E2A, E4Orf6, and VA. For example, functional recombinant AAV (rAAV) can be readily replicated and assembled in HEK293 cells (expressing endogenous E1A and E1B) via triple transfection of pHelper vectors expressing additional helper genes (E2A, E4Orf6 and VA), pRep/Cap vector encoding for Rep and Cap proteins, and AAV transfer vector with ITR-flanked transgene or gene of interest (GOI) (Figure 1
Recently, several groups reported the development of fixed-bed or single-use bioreactor based processes for large-scale AAV production via transient transfection. However, these pocesses were either based on adherent HEK293 cells or dependent on ultracentrifugation for the downstream processes [7–9]. Therefore, an efficient and fully scalable system (both upstream and downstream) for AAV production by transient transfection remained to be developed.
Several different naturally occuring serotypes of AAV are used in gene therapy, e.g., AAV1 for lipoprotein lipase deficiency, AAV2 for inherited retinal disease, and AAV8 and others for hemophilia [10–12]. These serotypes were chosen based on the target tissues. Additional and new serotypes were identified through search of human and nonhuman primate tissues, rational design, and directed evolution [13–15]. However, up to 70% of people have had prior exposure to some AAV serotypes and have thus developed pre-existing immunity (PEI) [16]. PEI renders the AAV treatment ineffective and excludes the majority of patients from clinical trials and therapies. In an effort to construct new AAV serotypes to reduce the impact of PEI, as well as to identify novel serotype that can target a variety of tissue types, Vandenberghe’s lab applied the ancestral sequence reconstruction (ASR) analysis to predict the putative novel AAV capsid sequences, which led to the isolation of a functional and potent AAV lineage, Anc80L65 [1]. The lineage has been the most extensively studied member of this Anc AAV library (~37,000 novel capsids) [17,18].
Here we show that we have established a productive and scalable manufacturing platform for Anc80 vectors using transient transfection in suspension HEK293 cells. We specifically isolated one ‘super-clone’ with productivities in the range of 2~3E+04 vg/cell for Anc80, comparable to or better than other available HEK293 suspension cell lines. An upstream process optimized for media and cell densities was successfully scaled up to 50L in single use bioreactors. Downstream processes using both capture and polishing columns resulted in highly pure full viral particles with potent infectivity in culture. Therefore, the high-production HEK293 manufacturing platform is an excellent option for the clinical and commercial production of Anc80 and other serotypes of AAV vectors.
Materials & methods
Single Cell Cloning
HEK293.3sus (Human Embryonic Kidney Cells) were obtained from ATCC (ATCC® CRL-1573.3™, Manassas, VA, USA). Commercially available HEK293 suspension cells (Life Technologies, Grand Island, NY, USA) were used as a control. HEK293.3sus cells were sorted using fluorescence-activated cell sorting (FACS, BD FACSAria II, San Jose, CA, USA) into two populations (GFP-high and -medium) based on the level of transfection efficiency using a plasmid encoding for GFP. The cell population displaying medium level transfection efficiency was chosen for limiting dilution into single cell in each well with 1% Penicillin-Streptomycin (Life Technologies, Grand Island, NY, USA) and 1% Human Serum Albumin (Octapharma, Lachen, Switzerland) in FreeStyle F17 medium (Life Technologies, Grand Island, NY, USA). Following plating, each well was screened for the presence of single cells under an optical microscope (Olympus, Tokyo, Japan) at 10x magnification. Single cells were monitored for cell growth on a daily basis. As colonies developed and cells in the wells started becoming confluent, the clones were transferred into larger low-adherence vessels until there were enough cells to be transferred into a shaker flask. 0.1% Pluronic-F68 (Life Technologies, Grand Island, NY, USA) was added to reduce shearing stress while shaking at 80 to 120 rpm in 37°C incubator with 8% CO2.
Medium Screening
In an effort to improve productivity, cell growth and productivity were assessed in several commercially available cell culture media. Cells were transferred into each medium to be screened and passaged to 5E5 viable cells/ml for at least 3 times prior to testing productivity. After three passages, cells were transfected in respective cell culture media and AAV titers were determined by qPCR.
Triple Transfection
Cells were seeded 1 day (shake flask) or 2–3 days (3 l and 50 l SUB) before transfection, to target achieving a cell density from 1E+06 to 3E+06 vc/ml at transfection. The plasmids used for the transfection were pHelper (Takara Bio, Mountain View, CA), pAAV-GFP (Cell Biolabs, San Diego, CA) and pRep/Cap for Anc80 (Vandenberghe lab, Boston, MA) or AAV8 (Synthesized by GenScript, Piscataway, NJ, USA). Briefly, the plasmids, PEIpro (Polyplus, Illkirch-Graffenstaden, France) or PEI MAX (Polysciences, Warrington, PA, USA) were diluted seperately in Opti-MEM medium (Life Technologies, Grand Island, NY, USA) and then combined. After 15 minutes incubation of PEI and plasmids mix, the complex containing solution was added to the cell culture for transfection. The culture was continued for 3 days before harvest.
AAV Production In 50 L Sub
A frozen vial of suspension HEK293 RCB cells was thawed and cultured in an appropriately sized Erlenmeyer flask (Corning, NY, USA) using FreeStyle F17 medium (Life Technologies, Grand Island, NY, USA) on a shaker in a humidified incubator at 37ºC and 8% CO2. The culture was subsequently passaged into appropriate size of shake flasks every 3 to 5 days until desired amount of cells were acheived to inoculate a 10 l N-1 culture using FreeStyle F17 medium in a 20 l WAVE bioreactor (GE Healthcare Life Sciences, WA, USA). Once the N-1 culture reached a desired cell density, it was used to inoculate a 50 l production culture at a target seeding density of 5E5 vc/ml in a 50 l SUB (Thermo Fisher Scientific, Waltham, MA, USA). The culture was sampled daily for cell count and nutrient analysis using Vi-CELL (Beckman Coulter, Indianapolis, IN, USA) and BioProfile FLEX (Nova, Waltham, MA, USA). Once reaching the desired cell density, the culture was transfected via triple transfection using PEI MAX and plasmids complex solution prepared in a WAVE bioreactor. Harvest was initiated 3 days post transfection.
Prior to the commencement of harvest, the culture was sampled for Anc80 AAV titer analysis. The culture was lysed using triton solution (Sigma-Aldrich, St. Louis, MO, USA) and treated with benzonase and magnesium chloride, and mixed with 500 mM sodium chloride prior to clarification. The culture lysate was then clarified using a depth filter and a 0.22 µm sterile filter (MilliporeSigma, Burlington, MA, USA). The clarified harvest was concentrated ~10 times and diafiltered for 5 diafiltration volumes (DV) by tangental flow filtration (TFF) (Repligen, Waltham, MA, USA). The resulting column load was stored at -80°C before purification.
Affinity Avb-Sepharose-Hp Column Purification Of 50 L Harvest
The purification runs were performed utilizing the AKTA Avant-150 chromatography purification system and UNICORN software. The column load generated and stored at -80°C was thawed in the 37°C water bath and filtered using the 0.22 µm Sartoguard PES filter. The filtered sample was loaded onto the VL22 column packed with AVB-Sepharose-HP (GE, Boston, MA, USA) resin at a linear flow rate of 150 cm/hr. The column was equilibrated with AVB Buffer A (50 mM Tris-base, 500 mM NaCl, 0.001% Pluronic-F68, pH8.0) prior to sample loading and washed with the same buffer after sample loading. The vectors were eluted using step elution with 100% AVB Buffer B (50 mM Sodium Citrate, 200 mM NaCl, 0.001% Pluronic-F68, pH3.5). Approximately 3CV of product eluate was collected into a clean bottle containing Neutralizing Buffer (220 mM HEPES, 440 mM Bis-Tris-Propane, 200 mM NaCl, 0.001% Pluronic-F68, pH9.0) to a neutralized product eluate.
Anion-Exchange Cimmultus-Qa Column Purification Of 50 L Harvest
The product eluate collected from the AVB-Sepharose-HP column was divided into two equal portions and each portion was purified using an 8 ml CIM-QA column (BIA Separations, Ajdovščina, Slovenia). The AVB column eluate was diluted with Dilution Buffer (20 mM Tris-base, 2 mM MgCl2, 0.001% Pluronic-F68, pH 8.0) to lower the sample’s conductivity to enable column binding. Prior to sample loading, the column was equilibrated with CIM-QA Buffer A (20 mM Bis-Tris-Propane, 20 mM NaCl, 0.001% Pluronic-F68, pH 9.0). The diluted sample was loaded onto the CIM-QA column at a flow rate of 1.5 CV/min and the column was then washed with CIM-QA Wash Buffer (20 mM Bis-Tris-Propane, 50 mM NaCl, 0.001% Pluronic-F68, pH 9.0). The vectors were eluted using step elution with 100% CIM-QA Buffer B (20 mM Bis-Tris-Propane, 200 mM NaCl, 0.001% Pluronic-F68, pH9.0). The product eluate was collected into a clean bottle. The purified eluates from the two CIM-QA runs were pooled and formulated into an appropriate buffer (PBS, 0.001% Pluronic-F68, pH 7.4) using the 100 kD, 115 cm2, MidiKros mPES Hollow Fiber TFF system (Repligen, Waltham, MA, USA). The formulated drug product was sterile filtered and stored at -80°C.
AAV Qpcr Titering
The cleared crude or purified samples were diluted 1:100 and further processed by DNase I (Promega, Madison, WI, USA) digestion at 37°C for 1 hour, followed by Proteinase K (Promega, Madison, WI, USA) at 37°C for 1 hour and at 95°C for 10 minutes. The treated samples were diluted 1:50 and the qPCR was set up with CMV-specific primers and probe. qPCR reactions were processed by LightCycler 480 II qPCR machine (Roche, Basel, Switzerland) using a thermal program as follows: 95°C for 5 minutes, 95°C for 15 seconds, 60°C for 1 minute; and repeated for 40 cycles. AAV titers were determined by comparison with a standard curve generated from linearized pAAV-GOI plasmids.
Flow Cytometry Analysis
One ml cell culture samples were collected into 1.7 ml Eppendorf tube, including negative control cells without transfection. Afer 5-minute centrifugation at 2000 rpm, cell pellets were gently re-suspended in 1 ml PBS. The percentages of GFP-positive cells were measured with BD Accuri C6 Plus (San Jose, CA, USA) following the standard operating procedures.
Silver Staining And Western Blot Analysis
AAV samples were prepared for electrophoresis under reducing conditions by adding 10µl of LDS 4X sample loading buffer (Life Technologies, Grand Island, NY, USA) to 4 µl of 10X sample reducing buffer (Life Technologies) and 24 µl of AAV. The samples were then denatured by heating for 10 minutes at 95ºC. Duplicate 4-12% BIS-TRIS SDS-PAGE gels (Life Technologies) were loaded at 1E10 vg/lane based on the viral titer as determined by qPCR assay. The gels were electrophoresed in 1xMES running buffer (prepared from 20x stock solution, Life Technologies) at 200V constant for 24 minutes. After electrophoresis one gel was analyzed by silver staining using Silver Quest Staining kit (Life Technologies) following the manufacturer’s recommended standard staining protocol. The gel was stained for approximately 8 minutes then the reaction was stopped and the gel was photographed. The second gel was blot transferred to a 0.45 µm PVDF blot using iBlot 2 dry blot transfer system in conjunction with an iBlot2 PVDF Mini Stack (Life Technologies). The resulting PVDF blot was probed with Western Breeze Chromogenic kit for mouse primary antibodies (Life Technologies) following the manufacturer’s protocol. The primary antibody used was an anti-AAV mouse monoclonal antibody to AAV viral capsid proteins VP1, VP2, and VP3 (American Research Products, Waltham, MA, USA) at a 1:1000 dilution. The chromogen was allowed to react on the blot for approximately 7 minutes after which the reaction was stopped and the resulting blot was dried then photographed.
Negative Staining For Transmission Electron Microscopy
The purified and formulated samples were sent to Texas A&M University (College Station, TX) Core Laboratory for negative staining and TEM analysis. Briefly, after thawing, samples were vortexed at level 4 for 3 seconds. 1.5 µl sample was added directly to carbon film and incubate for about 5 minutes without drying. The excess fluid was blotted with filter paper. The sample was rinsed three times on water droplets and blotted with filter paper and stained with 2% PTA or 0.2% UA with the grid tilted at 45 degrees. After blotting off excess stain, the sample was air dried and subjected to TEM analysis.
TCID50 Assay
HeRC32 cells were plated at 4E+04 vc in 50 µl per well in 96-well plate and incubated at 37°C in 5% CO2 incubator for 20 ± 4 hours. At the day of infection, 3.2 E+08 vp/ml human Adenovirus 5 Reference Material (ARM, ATCC, Manassas, VA, USA) was diluted in pre-warmed serum-free DMEM and used as diluent for AAV reference standard and AAV test article in a series of 10-fold dilution. After removal of medium in the 96-well plate, serially diluted AAV was added at 50 µl/well from Row A to G, Row H was used for the negative control. The set was repeated for Column 2 to10. Column 11 was used as an uninfected control. After incubation for 2 hours, pre-warmed complete DMEM was added at 50 µl/well and incubated for two days. The cells were lysed with 10 µl 10x lysis buffer (1% SDS, 10 mM Tris, 10 mM EDTA, 0.2 mg/ml Proteinase K, pH 8.0) at 37°C for 1 hour, 55°C for 1 hour and 95 °C for 10 minutes. The processed samples were diluted at 1:100 and quantified by TaqMan-qPCR following the procedures used for qPCR titering.
Results
Isolation Of Clonal Suspension Hek293 Cells With High Aav Productivity
The heterogeneous ATCC suspension HEK293 cells were used to isolate clonal cells for high AAV productivity. As the control Invitrogen clonal suspension HEK293 cells grow in FreeStyle 293 medium, we first adapted the ATCC cells into this growth medium until cell viability was consistently over 90% by Vi-Cell analysis. Next, we hypothesized that AAV productivity by triple transfection may positively correlate with cell transfection efficiency. Therefore, the ATCC cells were transfected with GFP-expressing plasmids and then subjected to cell sorting for GFP-positive transfected cells (Figure 2A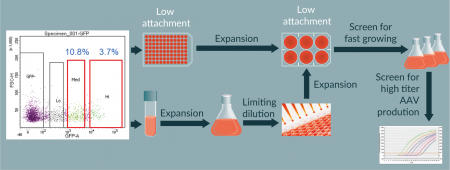
We identified 54 single cell clones from initial screening on Day 1 post plating. Among these clones, 10 sustained growth and developed into colonies (Figure 2B
Virus productivity of AAV/Anc80 in clone 5B8 was assessed and compared to a control commercially available suspension HEK293 using a triple transfection method. The average productivity of AAV/Anc80 in the 5B8 clonal cell line was determined to be 1.2E+10 vg/ml (n = 16) (Data not shown).
Optimization of AAV/Anc80 production in shake flasks
In the process of clonal HEK293 isolation, we started screening commercially available HEK293 cell culture media that support high titer AAV production using control suspension HEK293 cells. The initial screening of five media (Table 1) identified one medium (medium 3, FreeStyle F17) as the best medium for supporting both high cell density and high transfection efficiency. Medium 4 behaved similarly but is not cost efficient for larger scales. In order to further confirm the preliminary result and assess the growth of the 5B8 clone in additional media, we tested new medium 6 and 7 in addition to medium 3 and 5. Following triple transfection and qPCR titering for AAV/Anc80.GFP, we confirmed that Freestyle F17 remained the best medium for cell growth and AAV production by 5B8 cells (Table 1).
| Table 1. The screening of commercial media for suspension HEK293 cells based on supported cell density and transfection efficiency. ND, not determined. | |||||
|---|---|---|---|---|---|
| Medium type | Vendor | Supported cell density (vc/ml) | Supported transfection | Titer (vg/ml) | |
| Control | 5B8 | ||||
| 293 medium 1 | Gibco | 3–4 E6 | No | ND | ND |
| 293 medium 2 | Gibco | 3–4 E6 | Yes (80–90%) | 1.83E+09 | ND |
| 293 medium 3 | Gibco | 6–7 E6 | Yes (80–90%) | 1.56E+09 | 1.12E+10 |
| 293 medium 4 | Gibco | 1.5 E7 | Yes (80–90%) | 1.74E+09 | ND |
| 293 medium 5 | GE | 5–6 E6 | Yes (10–30%) | 1.39E+08 | 3.49E+09 |
| 293 medium 6 | GE | 5–6 E6 | No | ND | 3.97E+07 |
| 293 medium 7 | GE | 6–8 E6 | Yes (10–30%) | ND | 1.22E+09 |
To optimize the transfection process in suspension HEK293 cells, we evaluated the various transfection reagents. As shown in Table 2, we compared PEI MAX to PEIpro and Lipofectamine-LTX (Life Technologies). We observed an approximately 3-fold titer increase using PEI MAX compared to PEIpro. In the same experiment, the DNA amount was also varied and different plasmid ratios and different volume of diluent medium were assessed based on data obtained during previous studies [9]. Our data revealed a 2-fold increase in titer in the test condition compared to control.
| Table 2. Optimization of triple transfection for AAV production in suspension HEK293 cells. | ||||||
|---|---|---|---|---|---|---|
| Lipofectamine | PEIpro | PEI MAX | ||||
| Control | Test | Control | Test | Control | Test | |
| pHelper: pRC: pGOI (molar ratio) | 01:01:02 | 01:05.5 | 01:01:02 | 01:05.5 | 01:01:02 | 01:05.5 |
| DNA concentration (mg/ml) | 1 | 1.5 | 1 | 1.5 | 1 | 1.5 |
| Cocktail volume | 10% | 5% | 10% | 5% | 10% | 5% |
| Reagent: DNA | 03:01 | 02:01 | 02:01 | |||
| Cell density (vc/ml) | 1E6 | 1E6 | 1E6 | |||
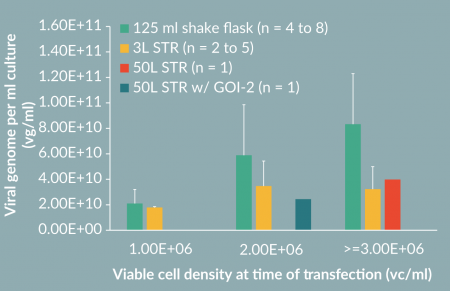
To determine if a higher cell density can result in increased AAV productivity, we seeded cultures at a different density at transfection. Accordingly, we increased the plasmids concentration proportionally, i.e. 3.2 µg/ml culture for 2E+06 vc/ml culture and 4.8 µg/ml for 3E+06 vc/ml culture. Interestingly, we successfully detected a proportional increase of AAV titer (Figure 5, 125 ml shake flask). Specifically, the productivity was increased up to 8E+10 vg/ml for AAV/Anc80.GFP at 3E+06 vc/ml cell density.
To test if the 5B8 clone cells could also support production of other AAV serotypes, we examined AAV8 production. The control HEK293 cells and 5B8 cells were triple transfected with AAV8 or Anc80 Rep/Cap plasmids at 1E+06 vc/ml cell density. FACS analysis on the GFP expression in these cells indicated that more than 80% of cells were transfected with each different serotypes in each cell lines, with 5B8 cells showing slightly higher GFP-positive populations (Figure 3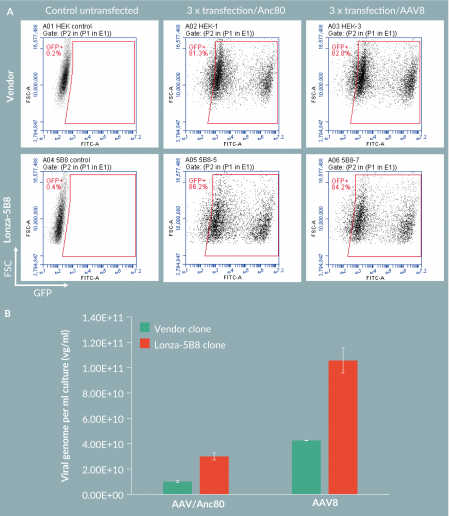
Upstream Scale-Up To 3 L And 50 L Single Use Bioreactors
To scale up the process from shake flasks to a 50L scale Stirred Tank Bioreactor (STR), we first tested the production in a 3L STR. Accordingly, we also tried increased cell density for AAV/Anc80 production in STR (Figure 5). We observed a nearly 2-fold increase of titer when culture was transfected at a cell density higher than 1E+06 vc/ml. To evaluate the AAV production process scalability, we performed two 50 l runs, one with pAAV-GFP and another with an undisclosed gene of interest (GOI-2). The complete process is presented inFigure 4
Downstream Purification Of Aav/Anc80 Vectors
Next, we purified and formulated AAV/Anc80.GFP vectors produced from the 50 l STR. We accomplished the purification by using a two-column purification system and formulated using the Hollow Fiber TFF system (Figure 4). We first captured the viral vectors from the column load (concentrated/diafiltrated clarified harvest) using an affinity AVB-Sepharose-HP column (Figure 6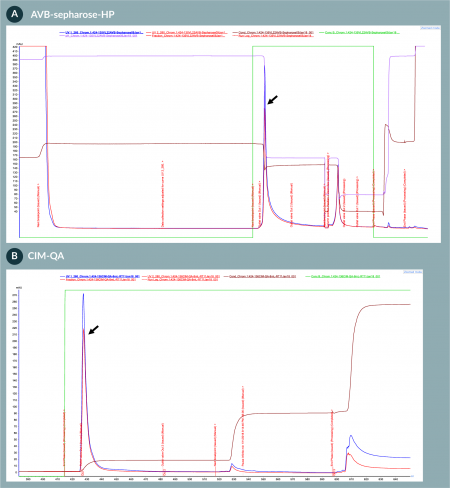
Analytics of purified AAV/Anc80 viral products
To examine the purity of the formulated AAV/Anc80.GFP vectors, we performed silver staining (Figure 7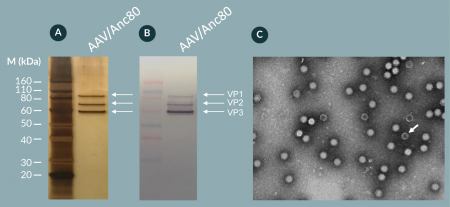
Discussion
Here we report a viable, cost efficient and scalable manufacturing platform for novel Anc80 and other AAV serotypes. The platform is based on upstream production by transient transfection of three plasmids in a highly productive clonal suspension HEK293 cell line in a single use bioreactor and downstream purification using a two-column chromatography process. With this platform, we have achieved the productivity of approximately 5E+14 purified and formulated Anc80 vector genomes from a 50 l production scale in about 8 weeks.
| Table 3. Summary of the analytical results of AAV/Anc80.GFP vector purification and formulation. | |||||
|---|---|---|---|---|---|
| Purification/formulation | Sample name | Titer (vg/ml) | Volume (ml) | Total (vg) | Step recovery (%) |
| AVB | Column load | 1.26E+11 | 3600 | 4.54E+14 | 100 |
| Eluate | 1.84E+12 | 127 | 2.34E+14 | 52 | |
| CIM-QA (2x runs) | Column load | 6.82E+10 | 1220 | 8.32E+13 | 100 |
| Eluate | 2.17E+12 | 42 | 9.11E+13 | 110 | |
| Formulation | Pre-formulate | 2.85E+12 | 78 | 2.22E+14 | 100 |
| Formulated | 1.18E+13 | 14.6 | 1.72E+14 | 77 | |
| Formulated/filtered DP | 1.01E+13 | 17 | 1.72E+14 | 77 | |
We isolated a clonal suspension HEK293 cells with superior AAV productivity using a triple transfection method. Compared to parental ATCC HEK293 cells, an isolated clone, 5B8, increased the AAV titers 5- to 10-fold (data not shown). This may be in part due to better transfection efficiency than the parental pool of cells, as the clone was selected based upon sorting for easily transfected cells. When we compared the 5B8 clone with control Invitrogen cells, both showed high and comparable transfection efficiency (over 80%). However, the 5B8 clone performed two to three times better than the control Invitrogen cells in AAV production for both Anc80 and AAV8. The 5B8 cell line was further characterized for stability, identity, bioburden and mycoplasma testing. A cGMP cell bank of clone 5B8 is in production and will be available through Lonza. Consequently, this high producer HEK293 clone provides an excellent platform for the clinical and commercial production of Anc80 and other serotypes of AAV.
We experienced some productivity loss during the scale up from shake flask to 3L and 50L STRs. This observation suggested the transfection mix prepared in WAVE bags for use in STRs was not as efficient as the transfection mix vortexed in tubes for use in shake flasks. Currently, we are working on the improvement of mixing of DNA and PEI for bioreactors.
Our downstream purification process is robust and scalable compared to traditional ultracentrifugation methods. The process involves a first step of affinity chromatography, followed by an anion-exchange polishing step. The virus yield following AVB-Sepharose column purification step was approximately 50%. This recovery rate is acceptable and has been reproducible at various scales. However, additional yield improvement studies will be conducted. In addition, we have tested other commercially available affinity resins but have not achieved reproducible results with different feedstreams over various runs. For the second column, a CIM-QA polishing column was used to further purify and enrich full viruses. Full capsid vector ratio of the final formulated product was initially 45% measured by AUC in the experiment. In the subsequent experiments with further improvements in the process, the percent of full capsid was enriched to higher level (up to 75%) by optimizing the chromatography elution step. Previously, Grieger et al. reported the purification of AAV from HEK293 cell lysate by iodixanol gradient followed by ion exchange chromatography [9], which posed a challenge for scale-up in GMP setting. Therefore, our two-column purification strategy provides a fully scalable and robust solution for AAV GMP purification processes.
While this process is a good near term solution, we are currently developing a transfection-free stable producer cell platform, which is expected to be a more robust, safe and cost-efficient process. The new stable cells would integrate necessary helper genes, Rep/Cap and GOI in the cell genome, allowing for large scale productions of AAV upon an addition of inducing agents, such as small chemicals, and thus eliminating the need of large amount of plasmids and tedious transfection steps. For now, transient transfection in a suspension format is a good bridging solution that provides a simple, fast and scalable process.
Financial & Competing Interests Disclosure
Financial support for this study was provided by Lonza. All authors of the manuscript are Lonza employees.
Acknowledgments
We thank Karen Magers and Daphne Lyonga for the critical reading of the manuscript and valuable suggestions.
References
1. Zinn E, Pacouret S, Khaychuk V et al. In Silico Reconstruction of the Viral Evolutionary Lineage Yields a Potent Gene Therapy Vector. Cell Reports 2015; 12, 10561068. CrossRef
2. Samulski RJ, Muzyczka N. AAV-Mediated Gene Therapy for Research and Therapeutic Purposes. Ann. Rev. Virol. 2014; 1, 427451. CrossRef
3. Robert MA, Chahal PS, Audy A et al. Manufacturing of recombinant adeno-associated viruses using mammalian expression platforms. Biotechnol. J. 2017; 12(3). CrossRef
4. Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus, J. Virology 1998; 72, 2224–32.
5. Grimm D, Kern A, Rittner K & Kleinschmidt JA. Novel tools for production and purification of recombinant adenoassociated virus vectors. Human Gene Therapy 1998; 9, 2745–60. CrossRef
6. Collaco RF, Cao, X & Trempe JP. A helper virus-free packaging system for recombinant adeno-associated virus vectors, Gene 1999; 238, 397–405. CrossRef
7. Chahal PS, Schulze E, Tran R, Montes J & Kamen AA. Production of adeno-associated virus (AAV) serotypes by transient transfection of HEK293 cell suspension cultures for gene delivery. J. Virological Methods 2014; 196, 163–73. CrossRef
8. Powers AD, Piras BA, Clark RK, Lockey TD & Meagher MM. Development and Optimization of AAV hFIX Particles by Transient Transfection in an iCELLis((R)) Fixed-Bed Bioreactor. Human Gene Therapy Methods 2016; 27, 112–21. CrossRef
9. Grieger JC, Soltys SM, Samulski RJ. Production of recombinant Adeno-associated virus vectors using suspension HEK293 cells and continuous harvest of vector from the culture media for GMP FIX and FLT1 clinical vector. Mol. Ther. 2016; 24(2), 297297. CrossRef
10. Bryant LM, Christopher DM, Giles AR, et al. Lessons learned from the clinical development and market authorization of Glybera. Human Gene Ther. Clin. Dev. 2013; 24, 55–64. CrossRef
11. Trapani I, Banfi S, Simonelli F, Surace EM & Auricchio A. Gene therapy of inherited retinal degenerations: prospects and challenges. Human Gene Therapy 2015; 26, 193–200. CrossRef
12. George LA. Hemophilia gene therapy comes of age. Blood Advances 2017; 1, 2591–9.
13. Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J & Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA 2002; 99, 11854–9. CrossRef
14. Zhong L, Li B, Mah CS et al. Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc. Natl. Acad. Sci. USA 2008; 105, 7827–32. CrossRef
15. Bartel MA, Weinstein JR, Schaffer DV. Directed evolution of novel adeno-associated viruses for therapeutic gene delivery. Gene Therapy 2012; 19, 694–700. CrossRef
16. Louis Jeune V, Joergensen JA, Hajjar RJ & Weber T. Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Human Gene Therapy Methods 2013; 24, 59–67. CrossRef
17. Landegger LD, Pan B, Askew C et al A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nature Biotechnol. 2017; 35, 280284. CrossRef
18. Pan B, Askew C, Galvin A et al. Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nature Biotechnol. 2017; 35, 264272. CrossRef
Affiliations
Bingnan Gu*
Research & Development, Lonza Houston Inc., Houston, TX, 77047, USA
Vijetha Bhat*
Research & Development, Lonza Houston Inc., Houston, TX, 77047, USA
Wenling Dong
Process Development, Lonza Houston Inc., Houston, TX, 77047, USA
Hai Pham
Process Development, Lonza Houston Inc., Houston, TX, 77047, USA
Stephanie Pubill
Process Development, Lonza Houston Inc., Houston, TX, 77047, USA
Nagarjun Kasaraneni
Research & Development, Lonza Houston Inc., Houston, TX, 77047, USA
Eric Onishi
Process Development, Lonza Houston Inc., Houston, TX, 77047, USA
Francesca Vitelli
Process Development, Lonza Houston Inc., Houston, TX, 77047, USA
Anandita Seth
Corresponding author:
Research & Development, Lonza Houston Inc. 14905 Kirby Dr., Houston, TX, 77047, USA
E-mail: anandita.seth@lonza.com
* Equal contributors.

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License</